
叔胺TOA从含钒钢渣直接酸浸液中萃钒除铁的研究
2021-08-18 07:29叶国华唐悦左琪胡渝杰陶媛媛陈子杨
矿产保护与利用 2021年3期
叶国华, 唐悦, 左琪,2, 胡渝杰, 陶媛媛, 陈子杨,2
1.昆明理工大学 国土资源工程学院,云南 昆明 650093; 2.省部共建复杂有色金属资源清洁利用国家重点实验室,云南 昆明 650093
引言
含钒钢渣产自于含钒铁水(钒钛磁铁矿经烧结和冶炼,钒进入铁水)的炼钢过程。含钒铁水的处理方法很多,包括吹炼钒渣法、含钒钢渣法、钠化渣法等。但无论采用哪种方法,都会有一定量的残钒进入半钢,并最终形成含钒钢渣[1,2]。
作为一类固废资源(冶炼渣),含钒钢渣来源广泛,总量也大,尽管其钒含量很低,仅1%~4%(以V2O5计,下同),但仍比石煤钒矿(0.3%~1.0%)要高一些,仍是很有价值的矿冶二次资源,可作为提钒的重要原料[1-3]。同时,国家《关于“十四五”大宗固体废弃物综合利用的指导意见》已明确提出,“鼓励从冶炼渣中回收稀有稀散稀贵金属等有价组分,提高矿产资源利用效率,保障国家资源安全,逐步提高冶炼渣综合利用率”。因此,在我国开展含钒钢渣提钒研究具有独特的资源基础和有力的政策支撑。
传统提钒主要采用焙烧后再浸出的工艺,但无论何种焙烧,都存在不同程度的废气污染,而且焙烧后再浸出,工艺复杂,不适于含钒钢渣的特点,钒转浸率很低[4,5]。针对传统工艺的不足,人们进一步提出了直接酸浸提钒的工艺[5-7],即在强酸条件下,取消焙烧工序直接用酸“浸取提钒”,它省去了焙烧环节,流程简化,作业环境好,并可获得理想的浸出率,已是提钒的发展方向[6-10]。但是,若不经焙烧直接酸浸,为提高浸出指标,往往须进行强酸浸出,此时由于酸度较高,酸浸过程基本失去了选择性,含钒钢渣中的很多组元都被溶解,所得到的直接酸浸液杂质很多(尤其是铁),净化富集十分困难,严重地影响了最终精钒产品的质量。因此,如何实现直接酸浸液的净化与富集(钒-铁分离是其主要任务),便成了含钒钢渣提钒的关键之一[11-13]。
含钒溶液的净化与富集,目前研究最多的是离子交换法和溶剂萃取法。对于离子交换,现在尚难找到合适的大容量树脂,而且该法对酸性溶液的效果较差[14];而溶剂萃取则具有容量大、适应性好、金属回收率高的优点,已成为生产高纯钒的有效方法。因此,一般而言,溶剂萃取法更适合于复杂酸性溶液(直接酸浸液即属于此)[15,16]。
萃取剂是溶剂萃取的基础和关键,适于复杂酸性溶液的萃取剂主要包括有机胺类(如N1923、TOA、Aliquat336)和酸性磷类(如D2EHPA、EHEHPA、Cyanex272)[15,17]。其中,酸性磷类为阳离子型萃取剂,可有效萃取金属阳离子,但除钒外,直接酸浸液中还含有很多的杂质阳离子,钒与某些杂质如Fe3+、Fe2+、Ti4+、Mn2+等均具有较高的分配比,因此酸性磷类的选择性较差,不太适合处理含金属阳离子较多的直接酸浸液[17]。有机胺类只能萃取在溶液中可以形成阴离子配合物的金属,因而选择性较好,除了钼、锆、铀以外(直接酸浸液中并不含这几种金属),其它金属包括铁很少能被其萃取,理论上钒与杂质组元的分离系数可达到103~104。因此,有机胺类更适合于处理含金属阳离子较多的直接酸浸液[18-20]。
目前,成功用于萃钒的有机胺类主要为伯胺、叔胺和季胺,且强酸性pH 1~2时叔胺萃钒能力一般优于伯胺、季胺,叔胺萃钒的分配比可达120~180。此外,与相近分子量的伯胺、季胺相比,叔胺水溶性小、萃取容量较大;更重要的是,叔胺的萃铁能力最弱,也即除铁能力最强[18-20],而含钒钢渣直接酸浸液中最主要的杂质即为铁,因此萃钒除铁宜优先考虑叔胺[18]。
鉴于上述分析,本文以叔胺TOA为萃取剂,采用溶剂萃取法对含钒钢渣的直接酸浸液进行净化与富集,考察了主要因素对萃钒除铁的影响,并对TOA的构效关系及其萃钒的溶液化学行为进行了分析,以期为业内提供重要参考。
1 试验
1.1 试样
直接酸浸液取自承钢集团含钒钢渣的不焙烧直接硫酸浸出工序。这一直接酸浸工序采用选择性分段浸出法(如图1所示),即浸出分两段进行,Ⅰ段稀酸预浸除杂(FeO、MgO全部浸出,钒不被浸出)、Ⅱ段强酸浸出提钒。

图1 直接酸浸——“选择性分段浸出”
Ⅰ段预浸除杂,在不损失钒的情况下,可预先抛除45%的铁;经Ⅰ段预浸除杂后,Ⅱ段再浸出提钒可获得相对纯净的含钒直接酸浸液,其中杂质铁的浓度较常规酸浸法明显降低。尽管如此,但由于含钒钢渣中铁含量基数很大,除Fe(Ⅱ)外还含有Fe(Ⅲ),而Ⅰ段浸出的只是Fe(Ⅱ),经Ⅰ段预浸除杂后,Fe(Ⅲ)不被浸出而与钒一起进入了Ⅱ段工序,导致含钒直接酸浸液仍含有很多的铁组元,Fe/V浓度比居高不下,净化与富集(钒-铁分离)的任务依然严峻。
直接酸浸液酸度很强(pH 0.6),其中的钒主要以+4、+5价形式存在。本研究选用叔胺TOA进行萃取,众所周知,TOA是一种阴离子交换型萃取剂,只能萃取阴离子,而强酸性溶液中,V(Ⅳ)一般是以阳离子(VO2+)形式存在,只有V(Ⅴ)才可以形成阴离子。因此,TOA萃取前需将直接酸浸液中的V(Ⅳ)氧化为V(Ⅴ),即萃取前需对直接酸浸液进行氧化预处理。本研究采用H2O2作氧化剂,钒氧化率达99%以上。
除了价态,溶液pH值对钒的离子形态亦有着重要的影响;只有pH>1.2时,V(Ⅴ)才绝对以阴离子形式存在(见图8)。而直接酸浸液pH值仅0.6,根据钒的溶液化学可知,在此酸度条件下,钒(氧化后为5价)仍主要以阳离子VO2+形式存在,同样不能被TOA所萃取。因此,萃取前还需对直接酸浸液进行中和预处理,调高其pH值,使钒(VO2+)全部转化为可被萃取的钒阴离子。
本研究以氨水作中和剂,直接酸浸液的pH值中和至不超过2.0,原因如下:浸出液中含有很多的杂质铁,氧化预处理后以Fe(Ⅲ)形式存在,由于Fe(Ⅲ)具有强烈的水解倾向,当加入氨水中和至溶液pH值高于2.0时,Fe(Ⅲ)便会开始水解生成沉淀,既不利于萃取操作,还会引起钒的共沉淀损失。因此,暂时确定将直接酸浸液中和至pH值1.7进行萃钒除铁研究。
氧化与中和预处理后的直接酸浸液作为萃取给料液,其化学组元列于表1。
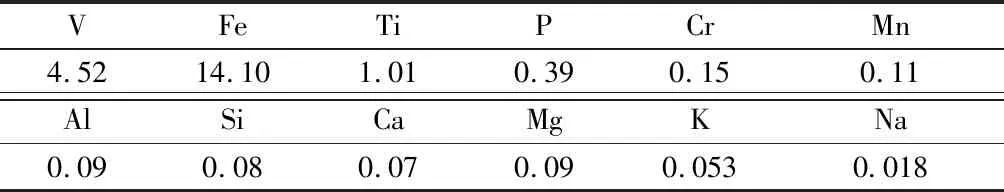
表1 给料液的化学组元 /(g·L-1)
由表1可知,给料液中的主要杂质组元为Fe、Ti、P、Cr、Mn;其中,Fe含量最高,Fe/V浓度比达3.12,萃取时应特别注意其共萃问题,尽可能地实现多萃V少萃Fe,即萃V除Fe进而实现V、Fe分离;Ca虽然是含钒钢渣中含量最高的组元,但在酸浸过程中它会转变成不溶的硫酸钙,不会进入酸浸液,因而给料液中Ca组元浓度很低,可基本不予考虑;含钒钢渣中Mg含量也较高,但经Ⅰ段预浸除杂后已基本被完全除去,故而给料液中Mg组元浓度非常低,同样不予考虑;含钒钢渣中Si(包括Al)也有一定含量,但Si、Al为酸性组元,很少能被硫酸溶出,故而给料液中Si、Al浓度同样很低,和上述杂质一样,不予考虑。
1.2 试剂
萃取有机相一般由萃取剂、改性剂和稀释剂组成。本研究选择叔胺TOA作萃取剂(使用前1mol/L H2SO4进行酸化处理)。然而,由于TOA在非极性稀释剂(如煤油)中的溶解度不太高,萃取含金属的配位酸根离子时,很容易产生第三相,妨碍两相分离。据报道,磷酸三丁酯(TBP)可作为改性剂,以抑制第三相的形成。我们的前期试验也证明了这一点,其结果表明,加入浓度5%体积分数的TBP即可很好地实现两相分离。该结果与文献报道一致:TBP对协同萃钒无明显作用,但显著影响两相分离,且TBP浓度选择5%体积分数为佳[21]。因此,选用TBP(5%体积分数)作为改性剂。此外,为防止出现“乳化”现象,选用磺化煤油作为稀释剂。
1.3 萃取方法
首先,使用稀硫酸或氨水,将水相(给料液,即预处理后的直接酸浸液,pH 1.7)的pH进一步微调至所需值,并混合不同体积的TOA、TBP、磺化煤油以制取有机相。然后,按照一定相比,将水相与有机相混合于分液漏斗中,在室温(23±2 ℃)下机械摇动至指定时间。分相后,用硫酸亚铁铵滴定法测定水相(萃余液)中的钒浓度,用重铬酸钾滴定法测定铁浓度,其它组元的浓度用电感耦合等离子体发射光谱法(ICP-OES)分析;负载有机相中的组元浓度根据质量平衡算出。萃取率E、分配比D、分离系数β分别由式(1)~(3)进行计算:
E=(1-CR/CF)×100%
(1)
D=Corg/CR
(2)
βV/Me=DV/DMe
(3)
式中:CF、CR、Corg分别为给料液、萃余液、负载有机相中组元的浓度,g/L;βV/Me为钒与某杂质组元的分离系数,下标V和Me分别代表钒和杂质组元。
2 结果与讨论
2.1 叔胺TOA浓度的影响
为确定合适的萃取剂浓度,考查了叔胺TOA浓度的影响,试验条件为:不同浓度(5%~25%)TOA+5%TBP+磺化煤油,水相初始pH 1.7,相比(A/O)为3,萃取时间5 min。结果如图2所示。

图2 TOA浓度对萃钒除铁的影响
从图2可以看出,开始阶段,随着有机相中TOA浓度从5%提高到15%,钒萃取率明显增加,而铁的共萃率一直维持在很低的水平;当TOA浓度为15%时,钒的单级萃取率达83%,而铁的共萃率仅3.3%;此后,继续增加TOA浓度从15%至25%,钒萃取率基本不变,而铁共萃率则显著增加,这显然对萃钒除铁不利。更为严重的是,当TOA浓度从15%增至25%,有机相黏度明显增加,有碍两相混合与分离。因此,为了实现两相的快速混合与分离,并尽可能在少萃铁的情况下多萃钒,TOA最佳浓度选择为15%,即有机相组成为15%TOA+5%TBP+80%磺化煤油。
由图2还可看出,铁的萃取率远远低于钒,此即意味着叔胺TOA选择性高,具有很强的萃钒能力,而对Fe的共萃能力则很弱。因此,本研究选择叔胺TOA作为萃取剂是可行的,它可以选择性地从直接酸浸液(往往都含有大量的铁杂质)中萃钒除铁。
2.2 初始水相pH的影响
控制溶液的pH值,使其中的钒以适宜的离子形式存在,对于萃钒而言十分重要。研究了初始水相pH值的影响,条件为:有机相15%TOA+5%TBP+80%磺化煤油,相比(A/O)为3,萃取时间5 min。结果见图3。

图3 初始水相pH值对萃钒除铁的影响
从图3可以发现,初始水相pH值是影响萃钒除铁的一个显著因素。当pH值小于1.8时,钒萃取率以及V/Fe分离比均随pH值的增大而快速上升;此后,pH值为1.8~1.9阶段,钒萃取率保持在87%以上,变化不大;进一步增大pH值至2.0及以上时,钒萃取率反而开始下降。而且试验发现,当pH≥2.0时,水相中的铁会开始水解(测试结果表明:给料液中95%的铁都是以易水解的Fe(Ⅲ)形式存在),生成胶状沉淀,吸附部分钒离子,妨碍两相分离并会造成钒的损失。由此得出,初始水相pH值选择1.8~1.9较为合适。
此外,研究表明[11],pH值主要是通过影响钒在水相中的离子形式(pH范围不同,钒的主导离子形式也不同),进而影响萃钒的。所以,为了更好地揭示pH值影响叔胺TOA萃钒的内因,采用核磁共振波谱(NMR)法对不同pH水相中钒的离子形式进行了测定,结果表明:(1)随着pH值从0增至4,钒的主导离子形式由[VO2]+(pH<0.8)逐渐变为[H2V10O28]4-(pH 0.8~3.6)、最后变为[HV10O28]5-(pH>3.6);(2)在最佳pH值范围内(pH 1.8~1.9,此时钒萃取率最高),水相中的钒几乎全部是以[H2V10O28]4-形式存在。这就说明,要想获得较好的TOA萃钒指标,应调节pH值、控制溶液中的钒以[H2V10O28]4-形式存在。
2.3 相比(A/O)的影响
考查了相比(A/O,水相与有机相体积之比)的影响,试验条件为:有机相为15%TOA+5%TBP+80%磺化煤油,初始水相pH 1.8,萃取时间5 min。结果示于图4。
由图4可以得出:(1)开始阶段,随着相比(A/O)的增加,铁共萃率降低明显,直至A/O为3;而在此A/O范围内(2.0~3.0),钒萃取率均较高,降低幅度较小。当相比(A/O)为3时,铁共萃率仅4.14%,而钒萃取率则高达87.53%,此时萃钒除铁效果最佳。(2)此后,随着相比(A/O)进一步增至3.5及以上,由于整个萃取体系中自由萃取剂的减少,钒萃取率开始明显下降。(3)而且试验发现,当相比(A/O)高于3时,由于负载有机相中萃合物的析出,两相界面间会出现少量污物,对萃取极为不利。对此现象的解释为:萃合物的极性很强,其在非极性溶剂煤油中的溶解度很小。当相比(A/O)较小时(<3.5),萃取饱和度不大,有机相中会有一定浓度的自由萃取剂,这部分自由萃取剂相当于极性改善剂的作用,增加了有机溶剂的极性,有利于萃合物在有机相中的溶解,故不会产生萃合物的析出;反之,当相比(A/O)较大时(≥3.5),萃取饱和度较大,有机相中自由萃取剂浓度减小,有机溶剂极性减小,其对萃合物的溶解性能下降,故会有萃合物析出。因此,为了萃钒除铁,并抑制界面污物的形成(即萃合物的析出),最佳相比(A/O)确定为3。

图4 相比(A/O)对萃钒除铁的影响
2.4 萃取时间的影响
一般而言,萃取时间越长,水相与有机相接触时间越长,萃取率越高,而萃取效率则会随之变差。考查了萃取时间的影响,试验条件为:不同萃取时间,有机相为15%TOA+5%TBP+80%磺化煤油,初始水相pH 1.8,相比(A/O)为3。结果见图5。

图5 萃取时间对萃钒除铁的影响
图5显示,随着萃取时间从1 min提高至3 min,钒萃取率可从71%明显提高至87%,而此时铁共萃率则变化很小,维持在4%以下。此后,继续延长萃取时间,钒萃取率几乎不再提高,而铁共萃率则在超过5 min时明显增加。这一现象表明,钒的萃取在3 min即可达到平衡,而对于铁,则需要很长时间方能达到平衡,铁的迟滞萃取性明显有利于萃钒除铁。另一方面,萃取时间长,分相时间也长,这会导致未来工业应用时生产周期长,生产效率低。因此,综上考虑,萃取时间选择3 min为宜。
2.5 最佳条件下的多级逆流萃取
为确定萃钒所需级数,保持两相总体积不变,以15%TOA+5%TBP+80%磺化煤油为有机相,对水相(初始pH 1.8)以不同相比萃取3 min,测出了当钒在两相中达到平衡后萃取等温线,如图6所示。

图6 钒萃取等温线
根据图6,利用McCabe-Thiele图解法,相比A/O为3时,要基本实现钒的全部萃取,所需理论级数为3级。实际结果与理论计算会有一定差距,在进行实际操作时,通常应在理论萃取级数的基础上增加1级,即实际级数选用4级。
所以,依据以上几个方面的研究和讨论,确定的最佳参数条件如下:萃取剂TOA浓度为15%,即有机相为15%TOA+5%TBP+80%磺化煤油;初始水相pH为1.8~1.9;相比A/O为3;萃取时间3 min;萃取级数4级。在最佳条件下,进行了4级逆流萃取模拟试验,结果见表2。
由表2可知,在Fe/V浓度比高达3.12时,最佳条件下经4级逆流萃取,98%的钒可被萃取,而铁则很少被共萃,萃取率仅4.4%;其它金属组元萃取率均很低:Ti低于2%,而Cr、Mn基本不被萃取;钒分配比达147,与之形成鲜明对比的是,其它金属组元的分配比均低于0.14,因而钒与金属组元的分离系数很高,达103以上,较高的分离系数说明,钒可很好地与直接酸浸液中共存的金属组元实现分离。最终,以TOA进行溶剂萃取,很好地实现了从含钒钢渣直接酸浸液中萃钒除铁(金属组元)的目标。这一结果亦充分证明,叔胺TOA具有良好的萃钒除铁(金属组元)性能。

表2 最佳条件下4级逆流萃取结果
但需要说明的是,非金属组元P的共萃率较高,后续洗涤、反萃时注意其踪迹走向,视情况作出除磷处理,以避免其对最终精钒产品质量造成不利影响。
3 TOA构效关系及其萃钒除铁的溶液化学行为
叔胺TOA属于中等强度的碱性萃取剂,化学式为(C8H17)3N,分子结构如图7所示。图7使用Materials Studio软件绘制,其中蓝色球为氮原子,灰色球为碳原子,白色球为氢原子。
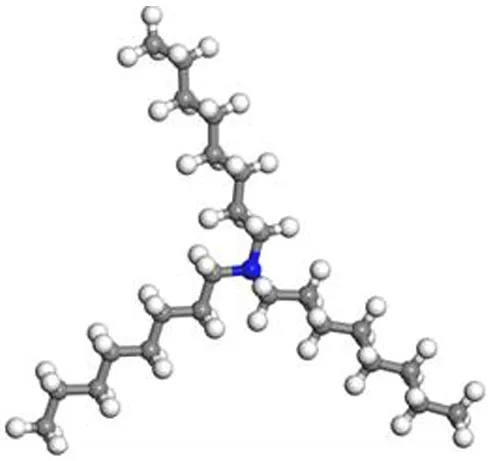
图7 TOA球棍模型
其中的N原子含有弧对电子,具有较强的给电子性,可与强酸作用生成胺盐阳离子(质子化),进而与水相中阴离子发生离子交换,萃合金属络阴离子,使阴离子进入有机相,与水相分离,完成萃取过程。
TOA使用前,往往需进行质子化处理,即硫酸酸化,将(C8H17)3N转化为[(C8H17)3NH]2SO4的形式:
2(C8H17)3N(O)+H2SO4→[(C8H17)3NH]2SO4(O)
(4)
对于含钒钢渣直接酸浸液,酸度很强、铁组元含量很高,其中,钒以两种价态存在,即V(Ⅳ)和V(Ⅴ)。作为阴离子型萃取剂,TOA只能萃取阴离子形式的V(Ⅴ),而强酸性溶液中,V(Ⅳ)主要是以阳离子VO2+的形式存在,难以被TOA萃取。因此,萃取前应进行氧化预处理以将V(Ⅳ)氧化为V(Ⅴ)。
H2O2是常用的氧化剂,其氧化性强、且不会引入新的杂质,因此,采用H2O2作为氧化剂,钒的氧化反应如下:
H2O2+2VO2+→2VO2++2H+
(5)
与此同时,直接酸浸液中的Fe(Ⅱ)亦被氧化为Fe(Ⅲ):
2Fe2++H2O2+2H+→2Fe3++2H2O
(6)
由式(5)~(6)可知,氧化反应有H+的生成及消耗,但总的来看,氧化预处理前后,浸出液pH基本不变,约为0.6。基于钒的溶液化学,在此pH下,氧化后的钒仍以阳离子VO2+的形式存在,同样不能被萃取。由图8可知,只有pH>1.2时,V(Ⅴ)才绝对以阴离子形式存在。因此,萃取前还应进行中和预处理,以调高浸出液pH值,使VO2+转化为可被TOA萃取的V(Ⅴ)阴离子形式。
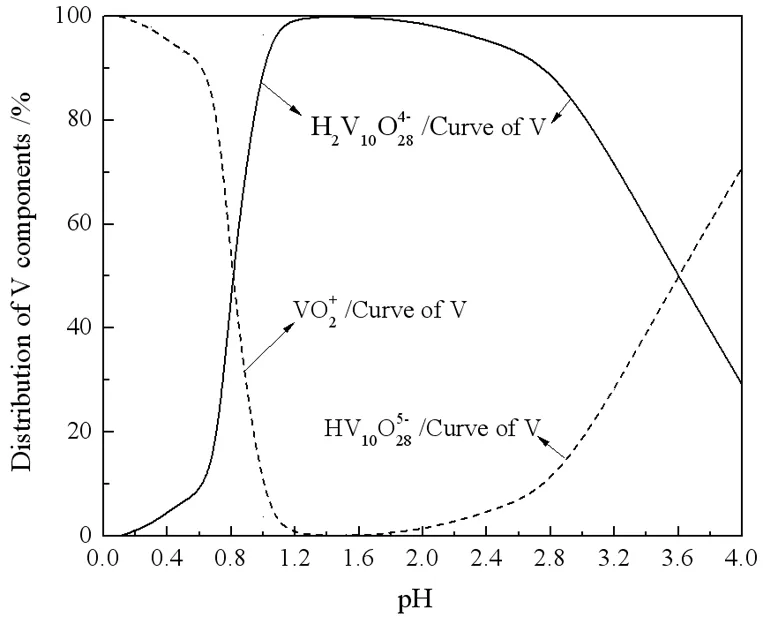
图8 酸性溶液中钒组元的分布系数
随着直接酸浸液中和时pH值的升高,VO2+可以与水聚合转化为多种形式的钒阴离子(即聚合钒氧根阴离子,通式可表示为[HxVyO(5y+x+z)/2]z-),如[H2V10O28]4-、[HV10O28]5-、[V10O28]6-等,这些钒阴离子均可被TOA萃取[12,13,18]:
[HxVyO(5y+x+z)/2]z-+z/2[(C8H17)3NH]2SO4(O)→
[(C8H17)3NH]zHxVyO(5y+x+z)/2(O)+z/2SO42-
(7)
其中,x、y分别为钒阴离子中氢原子和钒原子的个数,z为钒阴离子的价态的绝对值。
从式(7)可知,zmol TOA可萃取ymol钒,即1 mol TOA可萃取y/zmol钒,表明,y/z的值越大,钒的萃取量也就越大。所以,钒的萃取率与钒阴离子的具体形式有密切关系,为了保证较高的钒萃取率,应使浸出液中的钒以某种特定的阴离子形式存在。
在所有的钒阴离子中,十钒酸二氢根配阴离子([H2V10O28]4-)的y/z值最大,因此,当钒以[H2V10O28]4-的形式存在时,可获得最高的萃取率。此外,按照“最小电荷密度”原理,水相中金属离子或金属配合离子的电荷越少、离子半径越大,越容易被萃取。也即离子的“比电荷”(离子电荷与离子所含的原子数之比)越小,越容易被萃取。V(Ⅴ)阴离子的“比电荷”范围为0.1~0.6,[H2V10O28]4-的“比电荷”为0.1、最小,因此,[H2V10O28]4-比其他形式的V(Ⅴ)阴离子也更容易被萃取。
在强酸性溶液中,钒酸根离子主要存在形式有VO2+、[H2V10O28]4-、[HV10O28]5-,离子平衡如式(8)~(9):
10VO2++8H2O↔[H2V10O28]4-+14H+
(8)
[H2V10O28]4-↔[HV10O28]5-+H+
(9)
强酸性溶液中钒组元的分布系数如图8所示[13]。由该图中的曲线可知,当溶液pH为1.2~2.4时,V(Ⅴ)基本上完全以H2V10O284-的形式存在。也就是说,当pH值在1.2~2.4较宽范围内时,理论上而言,TOA萃钒都可以很好地进行。
此外,直接酸浸液中还存在着以下解离平衡:
HSO4-↔H++SO42-
(10)
pH值较小时,平衡左移,会导致HSO4-浓度过大,进而发生以下反应:
[(C8H17)3NH]2SO4(O)+2HSO4-→
2[(C8H17)3NH]·HSO4(O)+SO42-
(11)
这一反应会使[(C8H17)3NH]2SO4浓度减少,进而导致钒萃取率降低。增大pH值(即减少H+浓度),则有利于HSO4-的解离,减少它与[H2V10O28]4-的竞争,进而提高钒萃取率。
从这一角度而言,在pH 1.2~2.4的范围内,pH值越高,钒的萃取率也就越高。但试验发现,当pH≥2.0(高于1.9)时,溶液中的Fe开始发生水解沉淀,阻碍两相分离并引起钒的共沉淀损失。因此,溶液pH值不宜高于1.9。
对于杂质组元Fe(Ⅲ)而言,当pH<2.0时(视为强酸性溶液),基本上都是以Fe3+的形式存在,不会形成(络)阴离子,而作为一种阴离子交换型萃取剂,叔胺TOA是不能直接萃取Fe3+的。也有研究认为[13],Fe(Ⅲ)能与叔胺萃取剂发生交换反应的是其羟基络合物,而当pH<2.0时,Fe(Ⅲ)几乎不会形成羟基络合物。所以,可以认为在强酸性条件下(pH<2.0),叔胺TOA对Fe(Ⅲ)的萃取能力很弱,从而可达到萃钒除铁的目的。
4 结论
(1)用叔胺TOA从含钒钢渣直接酸浸液中萃钒除铁,最佳条件为:TOA浓度15%,即有机相为15%TOA+5%TBP+80%磺化煤油、初始水相pH 1.8~1.9、相比A/O为3、萃取时间3 min。TOA具有良好的萃钒除铁性能,在最佳条件下,经4级逆流萃取,钒萃取率达98%,而杂质铁则很少被共萃。
(2)叔胺TOA的N原子具有较强给电子性的弧对电子,经硫酸酸化,转化为[(C8H17)3NH]2SO4,可通过阴离子交换反应完成萃取过程。
经萃前氧化与中和,水相中的V(Ⅳ)氧化为V(Ⅴ),VO2+可转为多种形式的阴离子;当钒以[H2V10O28]4-特定形式存在时,可获得较高的萃取率,而且此时更容易被萃取;在pH 1.2~2.4较宽范围内,钒均可以[H2V10O28]4-的形式存在,而且在此范围内增大pH值,[H2V10O28]4-浓度以及HSO4-解离度增加,钒萃取率因而提高;但是当pH>1.9时,Fe(Ⅲ)会发生水解沉淀,阻碍两相分离并引起钒的共沉淀损失。
当pH<2.0时,Fe(Ⅲ)以Fe3+的形式存在,不会形成(络)阴离子或羟基络合物,难以被TOA共萃,从而可达到萃钒除铁的目的。
猜你喜欢
矿产综合利用(2022年3期)2022-11-25
西部交通科技(2022年7期)2022-10-22
今日农业(2022年14期)2022-09-15
今日农业(2022年13期)2022-09-15
中国药房(2022年7期)2022-04-14
西部交通科技(2020年6期)2020-03-01
当代陕西(2019年23期)2020-01-06
西部交通科技(2019年11期)2019-09-10
环球时报(2019-01-29)2019-01-29
科学与财富(2016年28期)2016-10-14
