
天山北麓典型水库病毒多样性及其中潜在病原体的分析
2021-09-24 11:49常娜娜地丽胡玛阿吉马正海毕玉海
环境科学研究 2021年9期
张 政, 常娜娜, 杜 菲, 地丽胡玛·阿吉, 张 成,2, 马正海*, 毕玉海,2
1.新疆大学生命科学与技术学院, 新疆生物资源基因工程重点实验室, 新疆 乌鲁木齐 830046 2.中国科学院微生物研究所,中国科学院病原微生物与免疫学重点实验室, 北京 100101
1989年Bergh等[1]发现,在湖泊中病毒粒子极为丰富,达2.5×108mL-1,该结果引起人们对海洋、湖泊、河流、水库等各类水环境病毒多样性研究的兴趣. 人们利用病毒宏基因组学分析发现,淡水环境中的优势病毒以噬菌体为主[2-4],这与噬菌体的宿主菌广泛分布于各类水环境中有关. 据报道,不同水环境中病毒群落组成和优势病毒种类存在差异,其主要受水质、水温、污染程度等环境因素,以及人类工农业活动和野生动物活动等因素影响,故水环境中病毒群落组成和多样性亦能反映水质状况,可作为水环境生态质量的评价指标[5-6].
天山北麓为冰雪消融及降雨汇集的山溪河流在天山北坡形成的淤积平原,具有独特的山地-绿洲-荒漠生态系统. 其中天山北麓中段位于准噶尔盆地南缘,东起乌鲁木齐市,西至乌苏市,南起前山带,北至沙漠边缘,地形总体由南东向北西倾斜,冰雪消融和降水补给形成乌鲁木齐河、玛纳斯河等河流,该区域是新疆维吾尔自治区工农业最发达和人口最集中的地区[7-9]. 天山北麓是干旱区山地生态系统和荒漠生态系统的交界地带,对自然环境敏感,其中水库等湿地尤为明显,稳定性和抗干扰性极差[10],生物多样性受自然条件和人类活动双重影响而变的极为复杂. 该研究选取天山北麓中段位于乌鲁木齐市城区内的三屯碑水库(STW)、五家渠市附近的八一水库(BYW)以及石河子市附近的蘑菇湖水库(MGW)作为典型样点,以病毒宏基因组学方法分析水库病毒群落的组成特征及其功能,以及其中潜在的致病性病毒,以期为该区域水库湿地的生态保护和合理化利用提供理论指导和参考依据.
1 研究方法
1.1 典型样点概述
STW(43°75′N、87°61′E)位于乌鲁木齐市南端的综合性游乐园水上乐园内,紧邻南郊客运站、地铁站和公交发车场等大型车流和人流聚集点,最大库容1.8×106m3,其水源由附近的乌拉泊水库及红雁池水库供给.
BYW(44°17′N、87°53′E)属于乌鲁木齐河流域,南侧及西侧紧靠乌鲁木齐市米东区羊毛工镇,东侧为甘莫公路,北靠新疆生产建设兵团102团团部,距五家渠市24 km,主要承担农业灌溉任务,正常蓄水位为462 m,库容3.0×107m3[11].
MGW(44°29′N、85°57′E)位于石河子市区西北18 km处,是玛纳斯河流域库容最大的水库,承担灌溉、养殖、调洪蓄水和纳污等功能,水库正常蓄水位为392 m,库容1.8×108m3[9].
1.2 试验材料和试剂
1.6 μm玻璃微纤维过滤膜购自英国Whatman公司;0.22 μm聚醚砜膜和0.8 μm聚碳酸酯膜均购自美国Millipore公司;DNase、RNaseⅠ、RNase H、Klenow exo-酶均购自美国赛默飞世尔有限公司;PrimeScriptTMcDNA第一链合成酶、PrimeSTAR PCR kit和MiniBEST DNA Fragment Purification Kit Ver.4.0均购自大连宝生物公司;QIAamp Viral RNA Mini Kit购于德国凯杰公司. 引物FR26RV-N (5′-GC CGGAGCTCTGCAGATATCNNNNNN-3′)和FR20RV(5′-GCCGGAGCTCTGCAGATATC-3′)由上海生工生物工程有限公司合成. 其他试剂均为国产分析纯.
1.3 水样采集
于2018年10月秋季枯水期末期采集STW、BYW和MGW表层水样,每个水库根据水域结构选6个采样点,采样点间隔500 m以上,每点采样850 mL,之后混合为 5 000 mL水样. 样品于-20 ℃便携式冰箱保存,当天运回实验室,存于-80 ℃冰箱备用.
1.4 病毒颗粒的富集
将Millipore快速过滤器与低压真空泵收集瓶连接,水样首先用1.6 μm Whatman GF/A玻璃微纤维膜过滤收集滤液,之后更换孔径为0.22 μm的聚醚砜滤膜再次过滤,收集滤液并向其中加入FeCl3充分混匀(铁离子终浓度达0.1 mg/L),室温黑暗条件下处理1 h,使病毒颗粒与Fe(OH)3形成絮凝物;随后用0.8 μm聚碳酸酯滤膜过滤,取出滤膜并剪成细条置于50 mL离心管中,加入5 mL病毒重悬缓冲液(1 mL MgCl2、1 mL EDTA、2 mL L-抗坏血酸,pH为6.5),于4 ℃黑暗条件下过夜;弃去滤膜,向病毒悬浮液中加入终浓度为17.5 g/L的NaCl以及8%的PEG 8 000,充分混匀后于4 ℃过夜;12 000g离心40 min,弃上清液获得病毒沉淀.
1.5 病毒核酸提取
病毒沉淀重悬于500 μL PBS,将重悬液均分4份置于1.5 mL离心管,每管加入7 μL Turbo DNA酶、2 μL RNA酶Ⅰ和15 μL 10×DNase reaction buffer,37 ℃水浴60 min;加入6 μL 0.5 mol/L EDTA,65 ℃水浴10 min,终止消化. 病毒核酸提取按QIAamp Viral RNA Mini Kit说明书进行,但在加入AVE病毒裂解液时不添加carrier RNA,以实现病毒DNA和RNA的同步提取[12].
1.6 病毒基因扩增
以上述病毒核酸为模板,FR26RV-N为引物,利用PrimeScriptTMⅡ 1st Strand cDNA Synthesis Kit合成cDNA第一链. 之后利用Klenow exo-酶合成ds cDNA,以病毒ds cDNA为模板,以FR26RV-N和FR20RV为引物进行PCR. 扩增参数:94 ℃,3 min; 94 ℃,30 s, 50 ℃,1 min, 72 ℃,1 min, 35个循环; 72 ℃,5 min. 用MiniBEST DNA Fragment Purification Kit纯化PCR产物,扩增的病毒基因产物在深圳市惠通生物科技有限公司测序.
1.7 病毒宏基因组学分析
经质控获得的病毒序列使用bbmap与NCBI nt数据库进行比对,除去宿主序列;使用Kraken基于序列不同k-mer长度的方法将序列片段与病毒数据库进行比对映射,剔除占比小于总病毒序列1%的序列,并对每个样本中的序列进行病毒分类注释;同时,用序列拼接软件SPAdes和MEGAHIT将序列组装成重叠群;使用BlastX对重叠群与Swiss-Prot蛋白数据库比对得到的序列进行功能注释.
1.8 柯萨奇病毒A10 (Coxsackievirus A10,CV-A10) VP1基因序列分析
基于高通量测序数据,使用SPAdes和MEGAHIT软件对3个水库潜在的致病性病毒做进一步分析,并对MGW样本中注释为肠道病毒A (EnterovirusA)的多个序列进行组装和拼接,将获得的CV-A10序列与BLATn序列进行比对,筛选并下载一致性较高和近年流行的CV-A10代表性序列. 使用ClustalW将获得的CV-A10VP1基因序列与下载的参考株序列进行比对,再通过MEGA 7.0软件采用Tamura 3-parameter model参数、bootstrap1000构建NJ系统进化树.
1.9 CV-A10 VP1抗原表位分析
使用DNAstar软件预测CV-A10VP1的抗原表位,以Chou-Fasman和Garnier-Robson两种方法预测其β-转角、β-折叠和α-螺旋等二级结构,以Kyte-Doolittle方法预测其亲水区,以Emini方法预测其表面可及性区域,以Karlus-Schuk方法预测其柔性区域,以Jamson-Wolf方法分析其抗原指数,根据文献[13]报道的方法综合分析VP1蛋白上述结构和功能特性,并预测其抗原表位.
2 结果与分析
2.1 病毒宏基因组测序结果概述
质控后,天山北麓中段典型水库STW、BYW和MGW中分别获得病毒序列 36 784 178、32 434 254 和 30 537 928 条,组装形成重叠群分别为 15 400 个(总长度为 14 764 455 bp,平均GC含量为47.03%)、11 894 个(总长度为 8 394 134 bp,平均GC含量为48.10%)和 30 771 个(总长度为 24 237 240 bp,平均GC含量为48.32%).
2.2 病毒群落组成与相对丰度分析
对各水库水样中病毒群落组成及相对丰度进行分析,科分类阶元结果(见图1)表明,3个水库病毒群落组成和优势病毒种类有一定共性,3个水库中相对丰度最高的优势病毒均属于有尾噬菌体目(Caudovirales),STW中长尾噬菌体科(Siphoviridae)的相对丰度达89.7%,BYW和MGW中肌尾噬菌体科(Myoviridae)的相对丰度分别为88.1%和66.0%. 同时,3个水库病毒组成也存在明显差异,STW病毒多样性较低,相对丰度较高的3个病毒科均属于噬菌体;另2个水库病毒种类相对丰富,除噬菌体占比较高外,还存在多种宿主病毒,如藻类DNA病毒科(Phycodnaviridae)、感染植物的帚状病毒科(Virgaviridae,亦称植物杆状病毒科)、南方菜豆花叶病毒科(Solemoviridae)以及感染动物的圆环病毒科(Circoviridae)和小RNA病毒科(Picornaviridae),MGW中尚有7.4%的病毒在科水平无法注释.
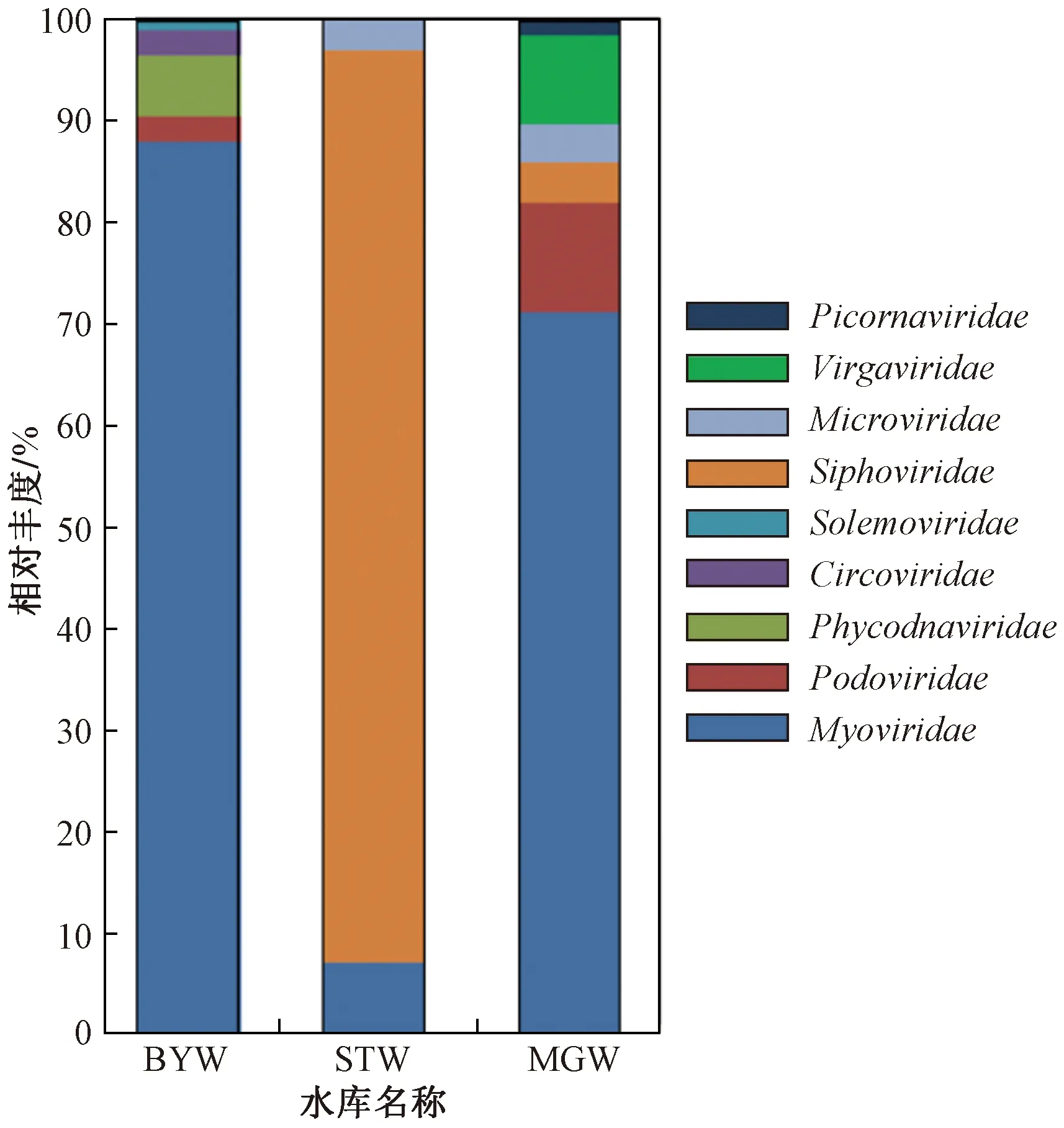
图1 水库病毒在科分类阶元的相对丰度Fig.1 The viral relative abundance of the reservoirs at family level
3个水库病毒在种分类阶元的结果如图2所示,MGW中相对丰度最高的病毒为欧文氏菌噬菌体ErwiniaphagevB_EamM-Y2和ErwiniaphageEa35-70,BYW中相对丰度最高的病毒为微囊藻噬菌体MicrocystisphageMaMV-DC和MicrocystisvirusMa-LMM01,STW中相对丰度最高的病毒为链霉菌噬菌体StreptomycesphageJay2Jay. 此外,3个水库中的噬菌体还包括气单胞菌属噬菌体(Aeromonasphage)、微小噬菌体科的Gokushovirinae、泛生菌噬菌体(Pantoeaphage)、沙门氏菌病毒(Salmonellavirus)、亚硫酸杆菌噬菌体(Sulfitobacterphage)、聚球藻噬菌体(Synechococcusphage)、浮丝藻噬菌体(Planktothrixphage)和噬蓝藻体(Cyanophage). 3个水库中还存在多种宿主病毒,如感染植物的黄瓜绿斑驳花叶病毒(Cucumbergreenmottlemosaicvirus)、辣椒轻斑驳病毒(Sowbanemosaicvirus)以及感染动物的类圆环病毒(Circovirus-likegenomeDCCV-7)和肠病毒(EnterovirusA).
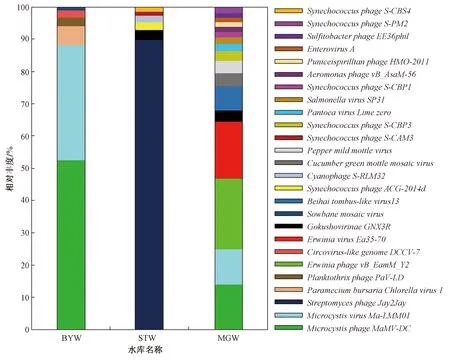
图2 水库病毒在种分类阶元的相对丰度Fig.2 The viral relative abundance of the reservoirs at species level
2.3 病毒功能基因组成与相对丰度分析
由图3可见,3个水库病毒功能基因组成相似,主要包括病毒结构蛋白基因、病毒包装相关基因和病毒复制相关酶的基因. 病毒结构蛋白基因包括病毒衣壳蛋白、尾蛋白等结构蛋白基因,其中衣壳蛋白基因相对丰度最高,在STW、MGW和BYW水库中分别达3.8%、5.6%和7.2%;病毒包装相关基因包括末端酶大亚基和内部支架蛋白基因等,末端酶大亚基在病毒包装中起主要作用,其基因在BYW、STW和MGW水库中的相对丰度分别为2.1%、2.8%和4.0%;病毒复制相关酶基因包括DNA聚合酶、解旋酶、依赖DNA的RNA聚合酶、依赖RNA的RNA聚合酶、HNH核酸酶等基因,DNA聚合酶基因在STW、BYW和MGW水库中的相对丰度分别为1.5%、3.2%和3.3%,解旋酶基因的相对丰度分别为1.6%、2.8%和3.3%.

图3 病毒功能基因的相对丰度Fig.3 The relative abundance of the viral functional genes
2.4 CV-A10 VP1基因序列分析
共获得CV-A10序列61条,得到一段注释为CoxsackievirusA10VP1的序列(简称“XJ201910”).VP1基因系统进化(见图4)显示:除作为外群的肠病毒CV-A12外,CV-A10VP1基因形成了6个进化分支,其中从MGW中获得的XJ201910和原始病毒株Kowalik聚为Clade A;Clade B主要包含2008年后从俄罗斯、法国、西班牙等欧洲国家分离的病毒株;Clade C主要包含2008年前从俄罗斯、格鲁吉亚、印度等欧亚国家分离的病毒株;Clade D主要为2010年前自我国山东省分离的病毒株;Clade E主要为2008年前自非洲分离的病毒株;Clade F包含2010年后自我国多个地区分离的病毒株.

图4 CV-A10 VP1基因系统进化树Fig.4 Phylogenetic tree of CV-A10 VP1 gene
2.5 CV-A10 VP1抗原表位分析
经预测,VP1蛋白包含12个β-折叠区,11个β-转角穿插其间;VP1蛋白还包含8个亲水性区域、7个表面可及性区域、11个柔性区域和11个抗原指数较高的区域. 综合分析以上潜在抗原表位的结构和功能区,预测VP1蛋白包含7个抗原表位,表1展示了7个预测表位的位置、序列以及相应区域的结构和功能特征.
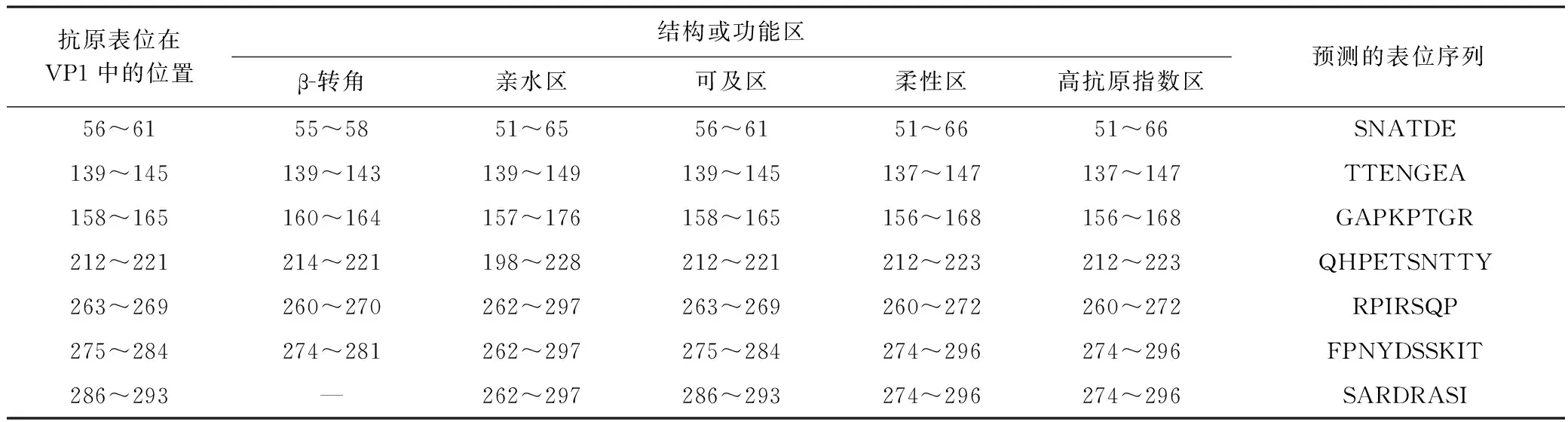
表1 CV-A10 VP1抗原表位预测
3 讨论
病毒宏基因组学最早用于研究表层海水,其中获得的病毒序列65%以上在现有数据库中没有相似的参考序列,这表明人们还缺乏对水环境中病毒群落的深入了解[14]. 目前,病毒宏基因组学在分析各类水环境病毒群落的多样性和功能及其与宿主关系和环境因素相互作用等方面得到了广泛应用[15],该研究使用病毒宏基因组学对天山北麓3个典型水库进行分析,以丰富天山北麓附近湿地的病毒学研究.
病毒颗粒的纯化和富集是获得足够核酸开展病毒宏基因组学研究的基础,超速离心、絮凝、过滤和PEG沉淀等方法是富集病毒颗粒和弃除宿主核酸的主要方法[16-19]. 其中FeCl3絮凝已经用于从海水、湖泊和库塘水样中富集病毒进行宏基因组学研究[16],与超速离心和超滤等方法相比,其所需水样较少. 该研究将FeCl3絮凝和PEG沉淀方法联用,成功地从5 L 淡水水样中富集病毒用于病毒宏基因组学研究,建立了从环境水样中富集病毒的简便高效的方法.
研究[4]显示,淡水环境中的病毒群落多以噬菌体为主,其与噬菌体的宿主菌在环境中广泛分布有关. 笔者研究显示,在科分类阶元各水库相对丰度最高的病毒均属于有尾噬菌体目,变形菌、拟杆菌和黄杆菌是有尾噬菌体目的主要宿主,富含以上菌类的江水和富营养淡水中有尾噬菌体的相对丰度较高[19-20]. 笔者所在课题组前期研究表明,变形菌、拟杆菌和黄杆菌为天山北麓水库中的优势菌,可作为有尾噬菌体的宿主,从而促使有尾噬菌体的复制和丰度增加. 同时,不同水库病毒群落的组成存在差异,BYW和MGW中肌尾噬菌体科的相对丰度较高,其为典型的烈性噬菌体,拥有广泛的宿主范围[21],在温带地区富营养淡水湖中的相对丰度在70%~80%之间[22];STW中长尾噬菌体科的相对丰度最高,其为温和噬菌体,是土壤中的优势病毒种类[3]. 因淡水环境中噬菌体宿主组成随水体营养、深度、位置以及周边环境和人为干扰等因素的影响而变化[23],从而影响其中噬菌体的种类和丰度. STW于2018年进行了淤泥清理工程,此工程可能促使淤泥中适于长尾噬菌体增殖的宿主菌带入水体并大量繁殖,进而为长尾噬菌体增殖提供了大量宿主. STW和MGW中微小噬菌体的相对丰度在3%以上,研究[24]显示其可通过周边陆地进入水体中.
在种分类阶元上,BYW中相对丰度最高的微囊藻噬菌体MicrocystisphageMaMV-DC和MicrocystisvirusMa-LMM01属于微小噬菌体科,这两种噬菌体宿主范围狭窄[25-26],而多数微小噬菌体科成员的宿主范围较广[27],基于这两种病毒的宿主专一性,可以推断水库中存在其宿主铜绿微囊藻和裂解微囊藻,而包括铜绿微囊藻在内的一些微囊藻可产生微囊藻毒素威胁人类健康[28]. STW中相对丰度最高的病毒为链霉菌噬菌体StreptomycesphageJay2Jay,该病毒前期自土壤链霉菌lividansJI1326中分离得到[29],Willoughby[30]曾提出部分链霉菌噬菌体更易感染湖底淤泥中的链霉菌,笔者结果再次表明STW的清淤工程促使淤泥中的宿主菌及其噬菌体进入水体并大量繁殖. 欧文氏菌噬菌体ErwiniaphagevB_EamM-Y2和ErwiniaphageEa35-70在MGW中相对丰度较高,这两种噬菌体均分离自梨树或苹果树下的土壤,其宿主为造成果树火疫病的欧文氏菌[31-32],表明欧文氏菌噬菌体丰度较高与库区农垦活动相关.
除上述丰富较高的噬菌体外,水库水样中还含有一定量的动物和植物病毒,其中包括多种病原体. BYW中圆环病毒科相对丰度为2.3%,其包含多种动物病原体[33],在种水平检测到Circovirus-likegenomeDCCV-7. 小RNA病毒科在MGW中的相对丰度为1.5%,该病毒科包括脊髓灰质炎病毒、口蹄疫病毒等多种感染人和动物的重要病原体,在种水平检测到的EnterovirusA是人类手足口病(hand and mouth disease,HFMD)的病原体,前期已有报道该病毒存在于水环境中并具有潜在公共卫生隐患[34]. 该研究从病毒宏基因组测序数据中挖掘到多个EnterovirusA基因序列,并获得了CV-A10VP1全长基因序列,CV-A10是引发HFMD的主要病原之一,其在不同地区的分离病毒株存在一定差异[35]. 分析表明,CV-A10VP1基因型与国内其他地区分离的病毒株差异较大(见图4),基于二级结构和功能域特征的综合分析预测CV-A10VP1蛋白包含7个抗原表位区,其中275~284和286~293位抗原表位与Zhu等[36]阐述的VP1蛋白在279~283和289位的B细胞抗原表位分布区域存在部分重叠,以上抗原表位可作为制备CV-A10疫苗以及诊断和治疗抗体的候选抗原表位. 感染植物的帚状病毒科在MGW中的相对丰度为8.1%,该病毒科包含烟草花叶病毒属、真菌传杆状病毒属等多种病毒[37],在种水平检测到感染植物的黄瓜绿斑驳花叶病毒、辣椒轻斑病毒和藜草花叶病毒,这些病毒均为植物的病原体. 其中,烟草花叶病毒属的辣椒轻斑病毒前期已从MGW周边种植的辣椒中分离获得,其还存在于健康人粪便中[38-39];另一植物病毒——南方菜豆花叶病毒在BYW中的相对丰度为1.1%,南方菜豆花叶病毒自然寄主为菜豆和豇豆,可导致花叶和斑驳等病害,在我国被列为检疫对象. 研究[40-41]发现,一些环境水样和污水样品中存在肠病毒、心病毒(Cardiovirus)和Cosavirus等致病性病毒,笔者所在课题组前期在MGW野鸟粪便样品中也分离到H1N1亚型禽流感病毒[42]. 该研究在水库水样中检测到多种可感染动物、植物以及人的致病性病毒,可能是库区农牧渔业和人类活动以及迁徙候鸟等野生动物活动所致,其对农牧业和公共卫生也构成了潜在的威胁.
天山北麓水库病毒群落的功能基因分析表明,3个水库中相对丰度最高的基因均为病毒衣壳蛋白基因,其次为尾蛋白和糖蛋白等结构蛋白基因,与富含有尾噬菌体的塔拉海洋病毒群落的功能蛋白基因组成一致[43]. 病毒包装基因以及DNA聚合酶等多种病毒复制相关酶基因的相对丰度均较高,说明水库病毒群落复制活跃. 研究[44]发现,HNH核酸酶促进了噬菌体基因组的复制,在笔者研究中病毒功能基因分析发现存在HNH核酸酶相关基因,推测病毒可能处于复制活跃期. 综上,3个水库中富含病毒包装和复制相关蛋白和酶的多种基因,表明水库病毒群落复制活跃.
4 结论
a) 该研究建立了FeCl3絮凝联合PEG沉淀从淡水中富集病毒的方法,利用病毒宏基因组学方法分析了天山北麓3个典型水库病毒的多样性及其功能.
b) 天山北麓3个典型水库的病毒群落均以噬菌体为主. 在科分类阶元上,BYW和MGW中肌尾噬菌体科的相对丰度最高,STW中长尾噬菌体科的相对丰度最高. 在种分类阶元上,STW、BYW和MGW中相对丰度最高的病毒分别为链霉菌噬菌体、微囊藻噬菌体和欧文氏菌噬菌体. 不同水库病毒群落组成差异与周边环境和人类活动等因素相关.
c) 水库病毒功能基因分析表明,噬菌体主要衣壳蛋白和尾蛋白等结构蛋白基因以及与病毒复制和装配相关蛋白编码基因的相对丰度较高,说明病毒复制活跃.
d) 水库中存在多种致病性病毒,包括感染植物的黄瓜绿斑驳花叶病毒和辣椒轻斑驳病毒,感染动物的类圆环病毒,以及感染人的肠病毒等,其中MGW中肠病毒相对丰度较高,并从中获得了HFMD的病原体CV-A10VP1全长基因,其与原始株Kowalik序列一致,说明水库中存在潜在致病性病毒.
猜你喜欢
传染病信息(2022年4期)2022-09-17
中国高原医学与生物学杂志(2022年3期)2022-06-22
昆明医科大学学报(2022年2期)2022-03-29
植物保护(2021年4期)2021-11-12
智慧健康(2021年2期)2021-03-15
科学(2020年3期)2020-11-26
当代水产(2020年3期)2020-06-15
科学24小时(2020年4期)2020-05-14
温州医科大学学报(2019年4期)2019-04-28
小星星·阅读100分(高年级)(2015年11期)2015-11-28
