
5-溴-2-(4-甲基哌啶-1-基)嘧啶的晶体结构及密度泛函理论研究
2021-12-15 07:54叶文君陈玉梅陈冬梅周志旭
人工晶体学报 2021年11期
叶文君,陈玉梅,陈冬梅,郭 倩,周志旭
(1.贵州大学药学院,贵阳 550025; 2.贵州省合成药物工程实验室,贵阳 550025)
0 引 言
含氮杂环化合物广泛存在于自然界和生物体内,因其高效低毒、环境友好,且具有独特的生物活性和结构多样性,在医药、农业、功能材料及化工等领域备受关注[1-4]。其中,嘧啶类衍生物具有广泛的生物及药理活性,如抗癌、抗病毒、抗菌、抗真菌、抗氧化和抗抑郁等[5-8]。此外,嘧啶类化合物在液晶、有机半导体、发光和非线性光学材料等科技领域也有进一步的研究价值[9-10]。作为研究广泛的C5位取代的嘧啶类衍生物的中间体,5-溴-2-(4-甲基哌啶-1-基)嘧啶(1)可通过Suzuki、Stille和Negishi等反应在C5位进行交叉偶联,并可被进一步修饰以开发新型嘧啶类衍生物[11-14]。
本文以5-溴-2-氯嘧啶为原料,首次报道了目标化合物5-溴-2-(4-甲基哌啶-1-基)嘧啶(1)的合成(见图1)。其结构经1H NMR、13C NMR、MS、FT-IR和X-ray等方法表征,并利用 Gaussian 09 软件包在B3LYP/6-311G(d,p)模式下对其最优构象进行了密度泛函理论(DFT)计算[15-16]。并对化合物1的晶体构象以及经DFT优化的结果与实验数据进行分析,揭示了标题化合物的部分理化性质,为进一步探寻该化合物的反应途径和应用研究打下基础。
1 实 验
1.1 主要仪器与试剂
IFS-55V 红外光谱仪(Bruker, Germany);Advance DMX400型核磁共振仪(Bruker,400 MHz,TMS为内标); APEX Ⅱ型单晶X射线衍射仪(Bruker);电化学工作站(上海辰华仪器有限公司);紫外可见分光光度计(上海元析仪器有限公司)。
文中实验所涉及试剂均为市售,并按照实验需求进行预处理。
1.2 5-溴-2-(4-甲基哌啶-1-基)嘧啶(1)的合成
在250 mL的两口瓶中,将5-溴-2-氯嘧啶(5.00 g,25.85 mmol)和碳酸钾(2.69 g,19.46 mmol)在搅拌下溶于乙腈(90 mL)。在25 ℃下将4-甲基哌啶(4.29 g,43.26 mmol)逐滴滴入反应混合液中,在25 ℃下搅拌反应8 h(TLC检测)。反应完成后,对反应混合物进行抽滤,滤液进行减压浓缩得到粗产物(6.75 g)。往粗产物中倒入冰水(100 mL),并在室温下进行搅拌3 h,然后对混合物进行抽滤。抽滤得到的滤饼用石油醚(10 mL)打浆得到化合物1(5.56 g,收率83.98%),为白色固体。1H NMR (600 MHz, CDCl3) δ 8.27 (s, 2H, 2-CH,4-CH), 4.70~4.58 (m, 2H,5-CH,9-CH), 2.87 (td,J=13.1, 2.7 Hz, 2H,5-CH,9-CH), 1.72 (d,J=13.5 Hz, 2H,6-CH,8-CH), 1.65 (tt,J=7.8, 5.3 Hz, 1H,7-CH), 1.16 (ddd,J=24.4, 12.6, 4.2 Hz, 2H,6-CH,8-CH), 0.98 (d,J=6.5 Hz, 3H,10-CH)。13C NMR (100 MHz, CDCl3) δ 159.83(Ar-C), 157.81 (Ar-C), 104.86 (Ar-C), 44.41 (C-N), 33.89 (-CH2-), 31.19 (-CH-), 21.91 (-CH3)。MS (ESI):m/z=257.12 [M+H]+。
1.3 X射线晶体结构测定
称取10 mg化合物1溶于5 mL乙腈中,溶液经有机尼龙针筒式过滤器(φ=0.22 μm)过滤转移至5 mL试剂瓶内。用聚乙烯薄膜包裹试剂瓶口,并在薄膜上扎三个小孔,静置;在室温下,溶剂自然对流并缓慢挥发后得到5-溴-2-(4-甲基哌啶-1-基)嘧啶(1)单晶。在显微镜观察下挑选透明、无裂痕、表面干净有光泽的晶体用于数据采集,所选晶体的尺寸为 0.4 mm×0.2 mm×0.2 mm。用Bruker APEX Ⅱ X衍射仪,以φ-ω扫描方式,用Mo-Kα射线(0.071 073 nm),在293 K下进行扫描和多次扫描吸收校正采集数据。衍射数据通过全矩阵最小二乘法修正后,再以SHELXL-2017/1程序精修,最后化合物1的晶体结构通过SHELXS-2017/1程序直接解出[17]。晶体数据如表1所示,最终的精修结果收敛于可靠性因子R=0.056 6和加权可靠性因子wR=0.168 8(w=1/[S2(Fo2)+(0.094 8P)2]),其中P=(Fo2+2Fc2)/3), (Δ/σ)max=0.000,S=0.923, (Δρ)max=492和 (Δρ)min=-92.9 e/nm3。CCDC: 1947031。
1.4 DFT计算
DFT 计算利用 Gaussian 09 软件包在 B3LYP/6-311G(d, p) 模式下进行,几何、电子和能量参数利用GuassView 5.0程序提取得到并解出晶体的最优构象[18-19]。
1.5 紫外-可见吸收光谱测试法和循环伏安测试
紫外可见分光光度计预热后,利用二氯甲烷进行基线校正,并对化合物1的二氯甲烷溶液进行紫外-可见吸收光谱测试,得到紫外-可见吸收光谱。以玻碳电极为工作电极、Ag/Ag+电极为参比电极、铂丝电极为对电极、四丁基六氟磷酸铵的二氯甲烷溶液(0.1 mol/L, 6 mL)为电解质在氮气环境下分别测得二茂铁(Fc, 6 mg)和化合物1(6 mg)的循环伏安曲线。
2 结果与讨论
2.1 晶体结构分析
化合物1晶体为正交晶系,空间群为P212121[晶胞尺寸为a=0.870 1(9) nm,b=1.081 9(1) nm,c=1.223 3(5) nm]。化合物1的晶体结构与 DFT优化的结构如图2所示。
由表2数据可知,通过对化合物1晶体学分析所获得的所有键长与键角都与DFT优化结构计算结果有良好的匹配度,并都在正常范围内。在Br(1)—C(1)、C(3)—N(3)和C(8)—C(10)所形成的平面两侧,分子内对应键长(如N(1)—C(3)和N(1)—C(2)、C(1)—C(4)和C(1)—C(2)、N(3)—C(5)和N(3)—C(9)等)和键角(如Br(1)—C(1)—C(4)和Br(1)—C(1)—C(2)、C(6)—C(7)—C(10)和C(10)—C(7)—C(8)、N(3)—C(5)—C(6)和N(3)—C(9)—C(8)等)都大致相等。这些数据表明5-溴-2-(4-甲基哌啶-1-基)嘧啶的分子结构在晶态下具有良好的对称性。在晶态下哌啶环以稳定构象椅式存在,且C(10)位上的甲基平伏着向环外延伸。
化合物1的晶胞堆积和氢键作用如图3所示,标题化合物中存在两个分子间C—H…π作用(C(7)—H(7)…π(嘧啶环)(0.281 0 nm)和(C(8)—H(8A)…π(嘧啶环)(0.296 8 nm))和两个分子内氢键(C(5)—H(5A)…N(2)(0.231 8 nm)和C(9)—H(9A)…N(3)(0.232 2 nm)),共同维系分子在空间上的稳定排列。

图3 (a)分子堆积图;(b)分子内氢键;(c)分子间氢键Fig.3 (a) Molecular stacking diagram; (b) intramolecular hydrogen bonds; (c) intermolecular hydrogen bonds
2.2 构象确定
分子的物理和化学性质与其构象有着重要联系,而可计算的构象分析能够帮助我们更好地理解有机化合物的物理化学性质,并阐明一些复杂的反应现象。因此,标题化合物的初始构象通过Spartan 08 程序对化合物1进行搜索和分析,对所有可能的构象异构体的几何构型再利用 Gaussian 09 软件包中的B3LYP/6-311G**模式进行优化和频率计算[20]。合并相同构象后得到两个相对稳定的构象异构体1-1(98.80%)和1-2(1.20%),如图4所示。两个构象异构体之间的区别主要是以哌啶环上甲基键型(直立键与平伏键)的不同产生的。化合物1的晶体结构与DFT优化后的基态结构的对比结果表明,构象1-1与化合物1的分子构象一致,且占据了98.80%。量子化学计算数据表明,化合物1的计算几何参数与X射线衍射数据大致相近,具有可靠性。
2.3 分子静电势
分子静电势(MEP)作为理论概念被广泛应用于分子识别、分子间的相互作用区域和作用力分析,可进一步探索分子的亲电、亲核和氢键位点。构象体1-1(与晶体结构相同)的分子静电势图通过使用B3LYP/6-311G (d, p)方法研究得到,静电势的定性大小以不同的颜色形式表现,静电势值按蓝、绿、黄、橙、红的顺序递减。如图5所示,正电性区域主要集中在4-甲基哌啶基和嘧啶环的氢原子上,电负性则体现在Br(1)、N(1)和N(2)上,是潜在的亲核攻击位点。

图4 DFT优化的化合物1的两种构象Fig.4 Two conformations of compound 1 by DFT-optimized
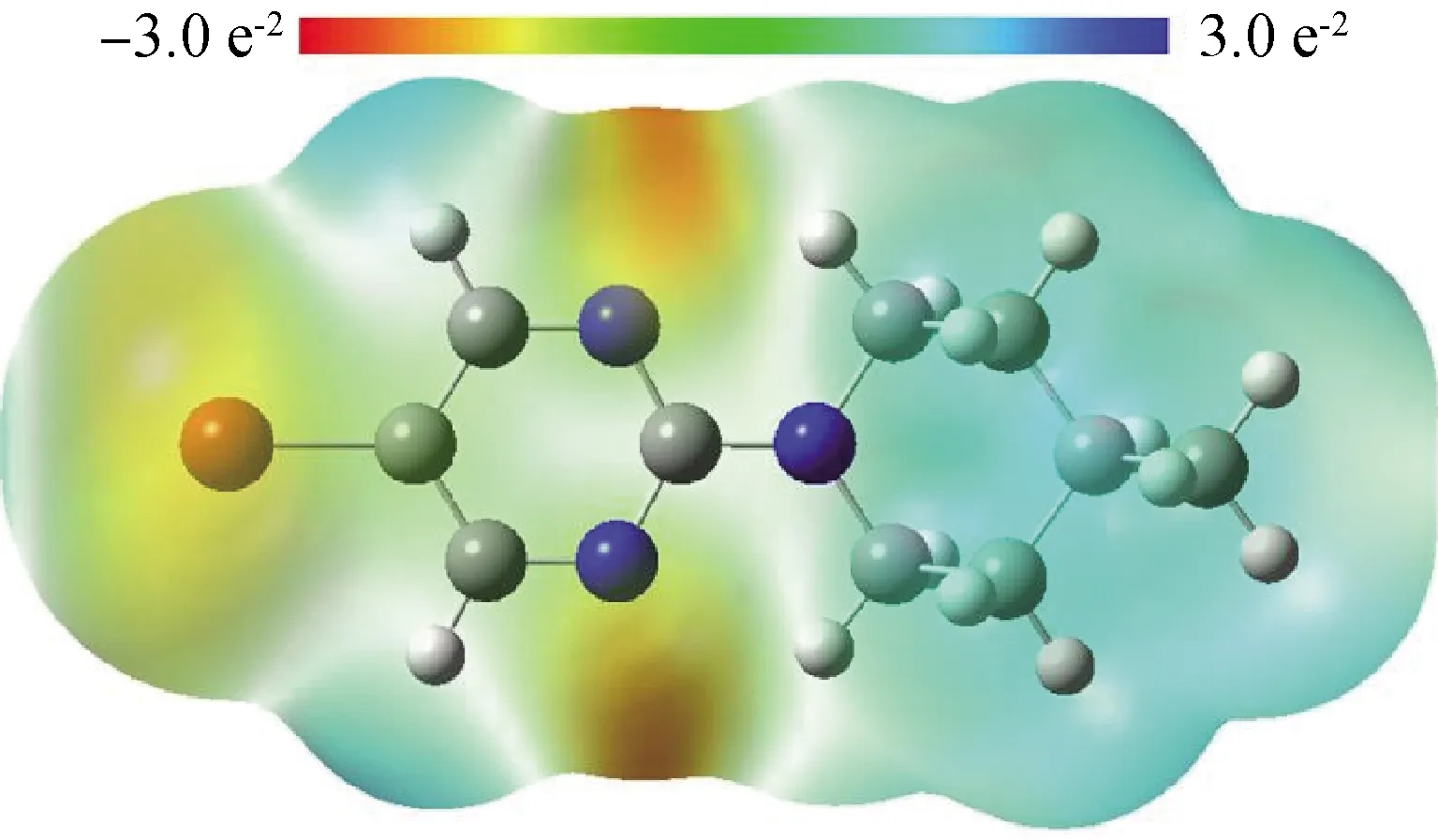
图5 构象体1-1的分子静电势图Fig.5 MEP map of conformer 1-1
2.4 前线分子轨道
作为参与分子内电荷转移的主要分子轨道,最高占据分子轨道(HOMO)和最低未占分子轨道(LUMO)决定着分子的电子得失、转移能力(即反应活性)和分子间反应的空间取向等重要化学性质。为了进一步研究构象体1-1(与晶体结构相同)的化学稳定性,使用了B3LYP/6-311G(d,p)方法计算了前线分子轨道能量(即EHOMO和ELUMO)及其轨道能带隙(ΔE)。图6中,红色和绿色分别代表正负区域,前线分子轨道能量EHOMO和ELUMO分别为-5.949 2 eV和-1.453 9 eV,轨道能带隙ΔE=-4.495 3 eV。对于计算的构象来说,得出的轨道能带隙值大意味着高激发态的激发能。因此,推测化合物1的电子不易发生跃迁且具有良好的化学稳定性和热力学稳定性。
采用循环伏安法与紫外-可见吸收光谱相结合的方法对化合物1的EHOMO,exp、ELUMO,exp和ΔEexp进行了测试,结果如图7所示。EHOMO,exp=-[E1OX-EFc/Fc+OX+4.8]=-[0.736-0.386+4.8]=-5.15 eV,带隙值ΔEexp=hc/λabs=1 240/382=3.246 eV(h为普朗克常数;c为光速),ELUMO,exp=EHOMO,exp+ΔEexp=-5.15+3.246=-1.904 eV。结果显示化合物1的电子不易发生跃迁,具有良好的化学稳定性和热力学稳定性。
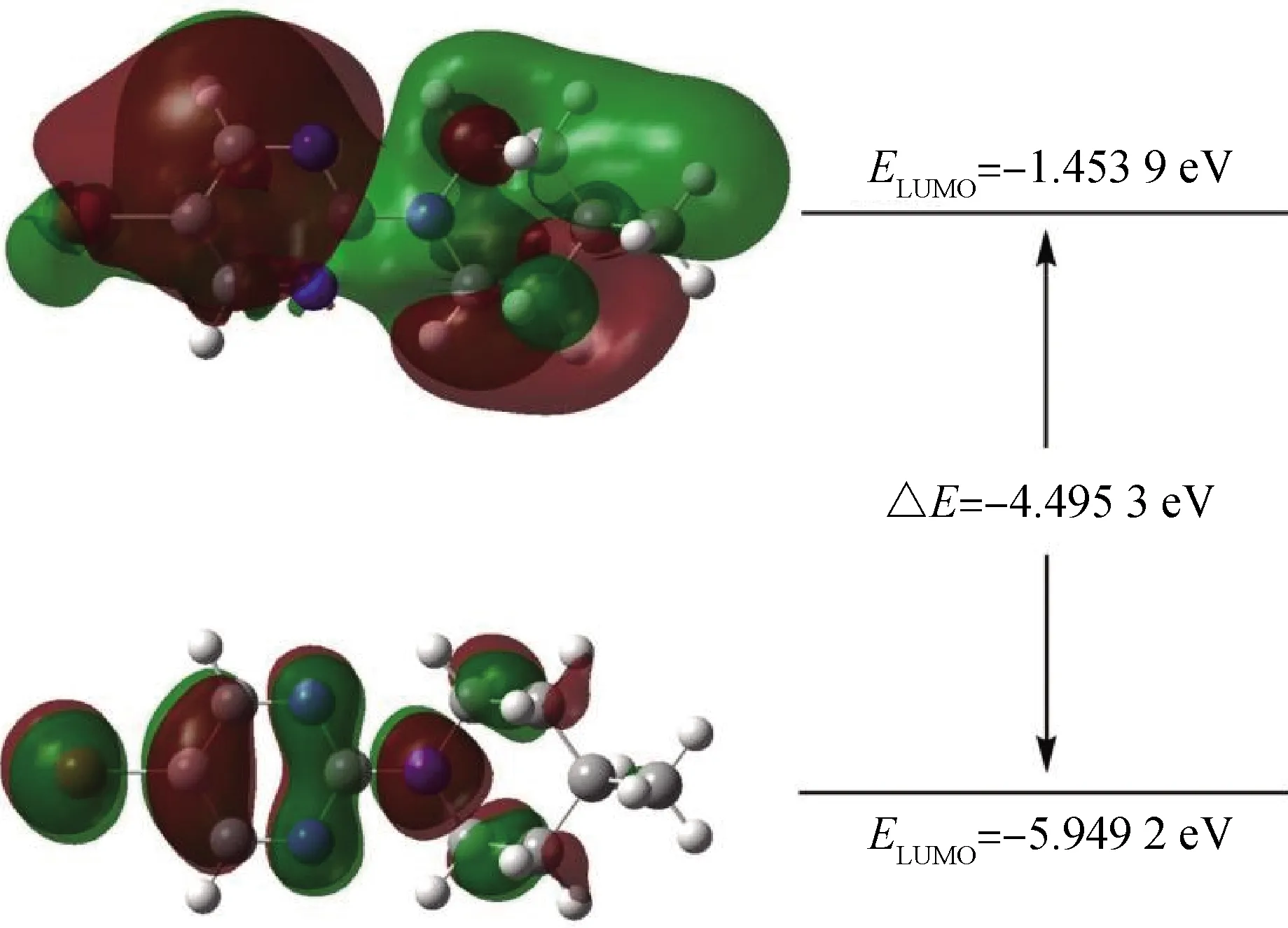
图6 构象1-1的HOMO、LUMO及轨道能隙带Fig.6 HOMO-LUMO and orbital energy gap of conformer 1-1

图7 (a)二茂铁的循环伏安曲线;(b)化合物1循环伏安曲线;(c)化合物1的紫外-可见吸收光谱Fig.7 (a) Cyclic voltammetry curve of ferrocene; (b) cyclic voltammetry curve of compound 1; (c) UV-visible light absorption spectrum of compound 1
3 结 论
本文通过一步芳香亲核取代反应得到了嘧啶类衍生物5-溴-2-(4-甲基哌啶-1-基)嘧啶,其结构通过光谱、核磁共振和质谱技术确证,并通过溶液结晶法得到单晶,进行X射线单晶衍射得到其晶体数据。并对其进行晶体学研究、DFT计算和构象分析,结果表明标题化合物的晶体结构主要由分子间和分子内的氢键作用来稳定晶体构象和形成晶体的三维结构,并且DFT计算参数与实验数据基本吻合,误差在合理范围内。计算结果与实验数据表明,标题化合物具有良好的化学稳定性的同时具有一定的反应活性。
猜你喜欢
分子催化(2022年1期)2022-11-02
石油化工(2022年9期)2022-10-19
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
智富时代(2019年7期)2019-08-16
智富时代(2019年7期)2019-08-16
山东青年(2019年3期)2019-07-21
农村农业农民·B版(2017年7期)2017-07-26
今日农药(2016年11期)2017-03-31
中学生数理化·高三版(2016年2期)2016-09-10
