
基于第三代纳米孔测序技术的茶树全长转录组分析
2022-01-07 02:26郭治友崔宝禄
福建农业学报 2021年10期
王 芬,木 仁,张 雯,马 媛,郭治友,崔宝禄,李 静
(黔南民族师范学院生物科学与农学院,贵州 都匀 558000)
0 引言
【研究意义】茶树Camellia sinensis(L.)O.Kuntze属山茶科、山茶属,异花授粉植物,起源于中国西南一带,为叶用经济作物,作为受大众欢迎的世界性饮品,具有非常强的保健作用[1-2]。茶产业作为中国的优势特色产业,在助力贵州农业农村乡村振兴方面具有重要的经济价值[3]。伴随着茶叶贸易全球化,消费者对茶叶质量的追求日益提高,提高茶叶品质是发展茶产业的重中之重。都匀毛尖茶在贵州省黔南州大面积推广,种植面积高达160万亩,40万群众以茶为业。都匀毛尖原料大部分为都匀本地种与引进的福鼎大白茶,福鼎大白茶发芽率高、抗旱性强,是十分优良的茶树遗传资源[4]。茶中的茶多酚、氨基酸和咖啡碱是茶叶滋味和品质的主要组成成分,其中茶多酚和氨基酸具有多种保健功能[5-7]。目前基于二代转录组测序技术对茶树的研究已经得到广泛应用[8-10],但由于其读长短,拼装困难,导致转录本的拼接组装不完整,而三代牛津纳米孔技术(Oxford Nanopore Technology, ONT)凭借其超长读长的优势可以弥补二代测序技术的不足,在转录本等信息的识别上更全面。基于ONT全长转录组测序可以从理论水平上更全面地挖掘与茶叶品质、独特芳香以及产量相关的代谢通路和生物过程。【前人研究进展】目前,单分子ONT技术已经在动物、植物、微生物等领域得到应用。Jansen等[11]利用ONT技术对欧洲鳗鲡的基因组进行了测序,与之前的草图相比准确性大大增加。Fellers等[12]通过ONT技术对感染小麦组织进行测序,证实了小麦条纹花叶病毒的存在,该结果表明ONT技术可以更准确地识别病原体。Giordano等[13]证明通过单分子ONT平台测序的数据足以完整地组装酿酒酵母S288C菌株。同时,一些茶树的基因组相继被测序,如云抗10号[14]、舒茶早[15-16]、小叶茶碧云[17]、古茶树[18]等,为改良茶叶品质、提高茶叶产量提供大量数据支持。RNA-Seq分析是解锁生命密码非常重要的工具,但由于第二代高通量测序平台[19]的转录组技术测得数据的拼接准确性不高,使得深入理解细胞生命活动困难重重。与二代技术相比,第三代测序技术具有通量高、读长长、成本低等优点,读长可达10 kb[20-21]。基于ONT[22]的全长转录组测序技术是一种单分子实时测序技术,并且在测序时无须打断RNA片段,所测即所得[23],大大缩减生物基因组重构和组装的时间和成本,为转录组学的研究减少阻碍。第三代测序技术的错误率虽然比二代高,大约为15%[24],但是利用更正软件加大测序深度可以大大降低错误率,可使准确率达到99.9%[25]。此外,庞丹丹等[26]利用PacBio三代测序技术对苦茶全长转录组进行分析,研究结果为探索苦茶特异性状相关基因标记的开发奠定基础。夏丽飞等[1]通过PacBio平台对紫娟茶树全长转录组进行分析,为开展紫娟茶树叶片呈色机理提供数据支持。【本研究切入点】目前,利用三代纳米孔测序技术研究茶叶的品质和滋味鲜有报道。【拟解决的关键问题】本研究利用三代测序平台ONT技术对都匀福鼎大白茶叶、根和茎进行全长转录组测序比较分析,旨在探究与茶叶品质相关的差异基因和代谢通路,为后续分子生物学研究提供数据参考。
1 材料与方法
1.1 试验材料
采摘贵州省都匀市黔南民族师范学院试验基地长势一致的福鼎大白茶扦插苗9株,分为3组,每组3盆,采摘嫩叶、嫩根和嫩茎分别作为叶、根和茎处理,每个组织3个生物学重复,共9个样本,包括叶片L1、L2、L3,根R1、R2、R3,茎S1、S2、S3,放入液氮中进行固样。将采集的样本放入干冰中送往北京百迈客生物科技有限公司进行全长转录组测序分析。
1.2 RNA提取、文库构建
利用北京天根生物技术有限公司的RNAprep Pure多糖多酚植物总RNA提取试剂盒(DP441)对都匀福鼎大白茶叶、根和茎的RNA进行提取。采用Nanodrop、Agilent2100及Agilent RNA 6000 Nano Kit对RNA的浓度和完整性进行检测,检测合格的RNA样品,使用oligod(T)磁珠从TotalRNA中纯化出poly(A)+RNA。其次利用Superscript Ⅳ reverse transcriptase反转录合成cDNA第一链,再使用带barcode的引物及LongAmpTaq2*Master Mix进行PCR扩增。然后使用NEBNext End repair/dA-tailing Module进行末端修复及加A。再次使用ONT SQKLSK109试剂盒及NEBNext Quick Ligationg Module进行测序接头的连接。最后使用PromethION测序仪及PromethION Flow Cells 9.4进行测序,并将三代转录组测序数据提交至NCBI-SRA数据库,BioProject的编号为PRJNA562747。
1.3 全长转录组测序
全长转录组测序平台使用Oxford Nanopore Technologies的PromethION,将原始下机序列中长度小于500 bp、Qscore小于7的低质量序列和核糖体RNA序列过滤掉,根据两端是否存在引物得到全长序列,对全长序列进行polish获得一致性序列,然后与安徽农业大学第一版的中国种茶树[27]基因组或构建的contig进行比对,将identity和coverage的值分别设置为0.9和0.85,去除冗余,得到转录本序列,再利用gffcompare v0.9.8将全长转录本与基因组已知的转录本进行比较,获得新基因和新转录本。随后进行SSR分析、ORF预测、转录因子分析、lncRNA分析,并且将所有的转录本、新转录本、新基因、开放阅读框、转录因子和LncRNA都上传到FigShare数据库,DOI号分别为:10.6084/m9.figshare.13671901;10.6084/m9.figshare.14370011。将Fold Change≥2且FDR<0.01作为筛选差异表达基因的标准,预测出叶与根、叶与茎、茎与根的差异转录本。最后,应用软件Blast2GO v2.5对差异转录本进行GO注释,并通过将转录本的蛋白序列和KEGG数据库中收录的蛋白序列进行BLAST比对得到KEGG功能注释信息,继而使用软件blast v2.2.31将转录本与kog202101数据库进行比对,得到转录本的KOG注释信息。叶与根、叶与茎、茎与根的差异转录本的GO、KEGG和KOG注释信息上传至figshare数据库,DOI号为:10.6084/m9.figshare.13671901。
1.4 转录本表达定量
应用CPM(Counts per million)[28]计算转录本表达量。
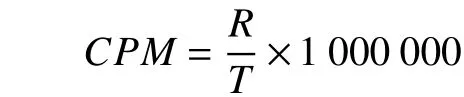
式中:R:比对到某一转录本上的reads数;T:比对到参考转录组的片段总数。
1.5 实时荧光定量PCR验证
为确保转录组数据质量准确性高,利用实时荧光定量PCR对随机选择的4个基因进行验证。试验材料和取样方法同1.1。利用Aidlab公司反转录试剂盒(TUREscript 1st Stand cDNA SYNTHESIS Kit)进行cDNA的合成。使用Primer5.0软件设计实时定量PCR引物(表1)。以茶树的GAPDH(GE651107.1 EST1434)基因为内参基因,利用2-△△Ct计算基因相对表达量,试验设置3个生物学重复。实时荧光定量PCR反应体系及程序按照Fermentas公司SYBR GREEN I说明书进行。

表1 qRT-PCR引物Table 1 qRT-PCR primer
2 结果与分析
通过Nanopore三代全长转录组测序对贵州省都匀市种植的福鼎大白茶叶、根和茎进行测序分析。每个样品测序产出clean data均达到7.93 GB,并且所有样本的平均质量值都达到Q9,9个样品得到的全长序列个数介于3768495~5078770(表2),利用经过polish处理的全长序列与第一个版本的中国种茶树进行minimap2.1.1[29]比对(表2),提取出69379个转录本。然后进行融合转录本的预测,9个样品的融合转录本个数为188~248。最后,预测出93102个SSR,获得7556个新基因位点,65795个新转录本,45852个ORF,6335个转录因子和2229个lncRNA,并完成了58398个新转录本的功能注释。

表2 Clean data数据Table 2 Clean data
2.1 测序数据分析
2.1.1 SSR分析 利用MISA1.0软件[30]对福鼎大白茶叶、根和茎的全长转录组做SSR检测,将转录本的序列长度≥500 bp作为筛选标准,共检测到50013条序列具有SSR位点,包含7种类型的SSR,共78931个。其中完美单碱基重复SSR个数为53433,数量最多,其次是完美双碱基重复,为27961个,然后依次是完美三、四、五、六碱基重复,数量分别为10316、586、254、552个。以搜索标准为1~6个碱基基序重复次数分别≥10、6、5、5、5、5在SSR位点中检测,单碱基重复出现频率最高是T/A(38139)。双碱基重复出现最多的是TC/GA(7356),其次是CT/AG(7226)。三碱基重复最多的是GAA/TTC(598)和CCA/TGG(581)。四碱基重复以TTTA/TAAA(70)和TTAT/ATAA(31)占优势。五碱基和六碱基重复频率最高的分别是TGTTA/TAACA(13)和GGTGCT/AGCACC(15)。以上研究结果与紫娟茶树[31]和藤茶[32]的结果基本一致,为未来开展茶树遗传图谱构建、SSR分子标记开发及培育良种提供理论基础。
2.1.2 新基因编码区序列预测 利用TransDecoder5.0.0(https://TransDecoder.sourceforge.net)预测出开放阅读框58355个,其中完整开放阅读框45852条。预测的完整开放阅读框编码蛋白序列长度范围主要在0~800氨基酸,0~100氨基酸有30455个,占52.19%,100~200个氨基酸的有22949个,占39.33%,200~300氨基酸有3936个,占6.74%(图1A),与云南金花茶[33]通过ESTScan预测的CDS长度大体趋势相似,以上结果表明转录组的序列质量较高。
2.1.3 转录因子预测 使用iTAK1.6软件[34]预测都匀福鼎大白茶转录因子,共预测到转录因子6335个,主要分为20类,其中GRAS家族转录因子数量最多,其次是MYB-related、RLK-Pelle_DLSV、WRKY和C3H(图1B),这些转录因子家族成员的获得为后续分子生物学的研究提理论参考。
2.1.4 LncRNA预 测 分 别 应 用Cpc[35]、Cnci[36]、Cpat1.2.2[37]、Pfam1.6[38]预测lncRNA,4种方法取交集共2229个(图1C)。lncRNA主要分为基因间lncRNA(LincRNA)(1878/84.3%)、反 义lncRNA(Antisense-lncRNA)(110/4.9%)、内 含 子lncRNA(Intronic lncRNA)(34/1.5%)、正义lncRNA(SenselncRNA)(207/9.3%),其中LincRNA最多,以上数据为将来研究lncRNA在茶中的调控机制提供重要参考。
2.2 功能注释
为了获得转录本的注释信息(表3),将得到的新转录本进行eggNOG、COG、NR、Pfam、Swissprot、KEGG、GO和KOG注释。

表3 新转录本注释Table 3 New isoform annotation
2.2.1 转录本表达量分析 根据CPM计算出转录本的表达量。采用CPM箱线图从整体上对9个样品的表达量进行比较,发现他们的表达水平基本上一致(图1D)。
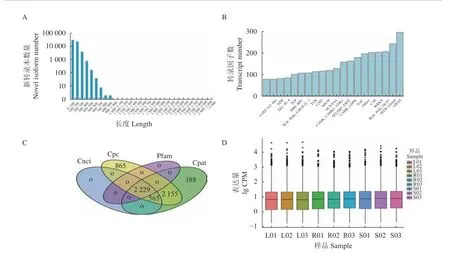
图1 开放阅读框、转录因子、长链非编码RNA和转录本表达量分析Fig. 1 Analyses on ORF, transcription factor, lncRNA, and transcript expression
2.2.2 差异表达转录本 利用DESeq1.18.0[39]进行叶、根和茎转录本的差异表达分析,筛选条件为Fold Chang≥2且FDR<0.01,叶和根的差异表达转录本最多,茎和根的差异表达转录本最少(表4)。对差异表达转录本进行数据库的功能注释,共注释了14306个差异表达转录本,分别有9649、2825和7349个转录本注释到GO、KEGG和KOG数据库中。

表4 差异表达转录本注释Table 4 Annotation of DETs
2.2.3 差异表达转录本GO注释 对差异转录本进行GO注释,一级分类主要包含3个类型,分别为生物过程、细胞组分和分子功能;二级分类主要分为52个类别。在叶与根的差异转录本中生物学过程涉及21个类别,以代谢过程(总转录本/差异转录本,29572/5019)、细胞过程(26938/4021)、单生物过程(19040/3581)最多。细胞组分包括16个功能组,其中细胞(27641/4454)、细胞部分(27478/4411)、膜(20620/3584)最多。分子功能分为15个功能类别,催化活性(30151/5408)、结合(24530/3744)、转运活性(3340/651)最多(图2)。叶与茎、茎与根的差异转录本的GO、KEGG和KOG注释信息上传至figshare数据库,DOI号为:10.6084/m9.figshare.13671901。研究结果丰富了都匀福鼎大白茶的分子生物学信息,可为进一步研究茶叶的品质提供理论参考。
2.2.4 差异表达转录本KEGG注释 KEGG数据库共注释到2825个叶与根的差异转录本,其中参与碳代谢(270、9.56%)、氨基酸生物合成(192、6.8%)、苯丙素生物合成(182、6.44%)、淀粉和蔗糖代谢(167、5.91%)、植物激素信号转导(137、4.85%)的差异转录本最多(图3)。参与茶叶品质形成中涉及茶叶滋味相关代谢途径的有氨基酸生物合成、各种氨基酸代谢、类黄酮生物合成和苯丙素生物合成。与香气相关的代谢途径有泛醌和其他萜烯醌生物合成和萜类骨架生物合成。研究结果为后期深入开展提高茶叶品质的研究提供理论基础。

图3 叶与根差异表达转录本KEGG功能分类Fig. 3 KEGG function annotation of DETs between leaf and root
2.2.5 差异转录本的KOG注释 叶与根的差异转录本与KOG数据库进行比对并根据其功能进行分类,有7349个转录本得到注释。按照功能一共分为25类,注释到一般功能预测(1531)中的差异转录本最多,其次是翻译后修饰、蛋白质转换、伴侣蛋白(924),次级代谢生物合成运输和分解代谢(812)。注释到KOG数据库最少的叶与根差异转录本数是染色质结构和动力学(30)、真核细胞的细胞外结构(20)、核结构(6)、细胞运动(0)(表5)。

表5 部分叶与根差异转录本的KOG分类Table 5 DETs KOG classification between leaf and root
2.3 差异表达转录本qRT-PCR分析
以茶树的GAPDH(GE651107.1 EST1434)基因为内参基因,随机选取4个差异表达基因(ONT.4041、ONT.8670、TEA019914、TEA007016)进行qRTPCR验证(图4),结果表明其表达量与转录组测序结果相一致,进一步说明转录组测序质量较高。
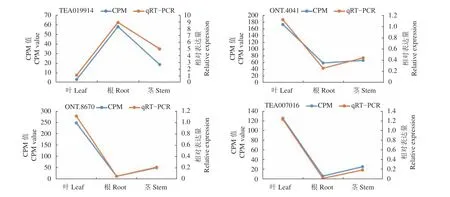
图4 转录组测序结果与qRT-PCR表达量比对Fig. 4 Comparison between RNA-seq and qRT-PCR
3 讨论
ONT第三代测序技术是近年来新兴的单分子测序技术[40],DNA/RNA链以一定的速率通过纳米孔通道蛋白时,单个碱基会引起不同电学信号的变化,根据电流信号对序列进行实时测定[41-42]。本文利用ONT全长转录组测序技术及生物信息学分析获得了福鼎大白茶叶、根和茎9个样品的全长转录组。通过CPM分析叶、根和茎9个样品的转录本表达量,发现它们的整体表达水平基本上一致。另外,我们共得到93102个SSR,其中完美单碱基重复和完美双碱基重复最多。单碱基、双碱基、三碱基和四碱基重复出现频率最高,分别是T/A、CT/AG、GAA/TTC、TTTA/TAAA,与朱兴正等[43]和鞠烨等[44]的研究结果基本一致,为未来开展茶树遗传图谱构建、SSR分子标记开发及培育良种提供理论基础。预测的45852个完整ORF序列,蛋白序列长度范围主要在0~800氨基酸,0~300个氨基酸最多,与夏丽飞等[1]和潘敏等[45]的研究结果大体相似,表明转录组的序列质量较高。我们还对转录因子、LncRNA进行了分析,研究结果为将来研究LncRNA在茶中的调控机制提供重要参考,为后续分子生物学的研究提供理论参考。
根据GO功能显著性富集分析,上调基因和下调基因在生物过程中,代谢过程(2162/2857),即叶与根中有2162差异转录本在生物过程中上调,2857个转录本在此过程中下调。单生物过程(1688/1893),细胞过程(1673/2348)、生物调节(1673/633)等显著富集。在细胞组分中,细胞(1623/2831)、细胞部分(1608/2803)、膜(1597/1987)、膜部分(978/1201)等显著富集。在分子功能中,催化活性(2496/2912)、结合性(1771/1973)、转运活性(331/320)等显著富集。以上结果是叶与根差异转录本数最多的生物过程,朱兴正[43]等采用PacBio平台分析了保护品种云茶1号的全长转录组,Unigene的GO功能注释与本研究结果相一致。同时,以上结果显示在这些生物过程中大部分下调转录本数都比上调转录本多,即在都匀福鼎大白茶树生长发育过程中,参与叶生物过程的转录本比根多。
在KEGG中,叶与根的差异转录本中有182个差异基因参与苯丙素的生物合成,KEGG直系同源基因ID号为ko00940。对该通路的苯丙氨酸、络氨酸、色氨酸的生物合成到木质素的通路进行深入分析,涉及6个节点,其中4个节点中的基因全部上调,2个节点既有上调又有下调基因。将涉及的所有基因根据基因表达量进行聚类分析,TEA019411、TEA017242、TEA028682、 TEA029025、TEA006839、TEA017067、ONT.2421、TEA008747、TEA031671、TEA005749等基因在叶中高表达,都属于K00430,并且亲缘关系较近,与过氧化物酶有关,参与氧化应激反应。过氧化物酶与光合作用、呼吸作用有关,并能使组织中所含的某些碳水化合物转化成木质素,增加木质化的程度,是组织老化的一种标志。在氨基酸生物合成中,有192个差异基因参与,其中涉及谷氨酸合成酶的基因有ONT.24127.2和TEA003892.1等,并且这两个差异基因在根中表达上调。茶氨酸是在茶树根部由乙胺和谷氨酸在酶的催化下合成的,然后运输到叶部,参与代谢过程。而茶氨酸的降解与阳光有关,影响茶氨酸向儿茶素转化,因此,茶氨酸的合成与降解与茶叶品质密切相关。以上结果表明,茶树叶片通过光合作用等一系列反应形成与茶叶品质相关的化合物。
叶与根上调和下调差异转录本KOG分类中,一般功能预测(611/920),上调转录本611,下调转录本920。翻译后修饰、蛋白质转换、伴侣蛋白(349/575),碳水化合物的运输和代谢(339/405)的分类中,大部分下调转录本比上调转录本多,再一次说明茶树生长过程中叶的重要性。本研究的全长转录组数据和各项结果丰富了茶树的分子生物学信息,可为进一步研究茶叶的品质和独特的芳香及与之相关的基因提供理论基础。为保障茶叶产量和品质打下良好基础,为后续良种选育及分子生物学研究提供重要的数据支持。
猜你喜欢
东坡赤壁诗词(2022年3期)2022-05-29
茶道(2022年3期)2022-04-27
云南画报(2021年10期)2021-11-24
科学之谜(2021年2期)2021-04-25
科学导报(2020年54期)2020-09-09
茶道(2020年4期)2020-08-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
农产品市场周刊(2017年18期)2017-06-07
延河·绿色文学(2016年8期)2016-05-14
