
第一性原理研究3C-SiC/Mg复合材料界面的电子结构
2022-03-04 02:32尧军平唐锦旗陈致君
原子与分子物理学报 2022年4期
吕 昭,尧军平,唐锦旗,陈致君
( 南昌航空大学 航空制造工程学院,南昌 330063)
1 引 言
镁合金有着导电导热性能良好、刚性好、密度低等特点,关于镁合金的应用、研发备受各界关注,但镁合金由于耐蚀性能差、强度低、易燃性等缺点使得镁合金不能满足人类在更多场合实际应用的需要,由此对镁合金新型材料的研发制备迫在眉睫.镁基复合材料拥有高比强度、高比钢度、高比模量等特性,被认为是21 世纪高新技术产业中最有希望被大量利用的轻金属材料之一,广泛的应用在汽车制造、电子设备、航空航天、国防装备等领域[1-4].
界面的结合性能对于复合材料有着重要的影响[5],DFT( Density functional theory) 在界面性能的研究方面有突出贡献,能较为直观的给出界面处原子位置,而且可以在电子级别分析界面的结合机制[6,7].而近年来计算机技术的飞速发展,计算机模拟技术逐渐在材料的开发研究中变得重要起来,材料的研发模式开始由传统的实验方式逐步转为理论模拟.其中第一性原理的计算方法又是其中最为可靠的.例如,Shingo 等人[8]对3C-SiC(111) /Al 界面进行了第一性原理赝势计算,3C-SiC(111) 的C 终端界面的结合能大于Si 终端界面的结合能.Liu R 等人[9]研究了Mg( 0001) /TiB2(0001) 界面的界面结构,发现Mg 原子与Ti端TiB2表面和B 端TiB2表面结合时,分别形成金属/共价混合键和离子键; Mg/TiB2界面的界面能远大于α - Mg/Mg 界面.Wu 等人[10]研究了Al(111) /6H-SiC(0001) 的界面结合及沿Z 轴拉伸时的界面断裂特性,结果表明,在Al( 111) /6H-SiC(0001) 界面,C 端接界面的粘附功为2.689 J/m2,高于端接界面的1.649 J/m2.在抗拉强度方面,C 端接界面高于Si 端接界面.两种复合材料的强度均高于纯铝,但延展性较弱.熊辉辉等人[11]用第一性原理方法计算了6 种不同Ti(0001) /TiB2( 0001) 界面的黏附功和界面能,研究结果表明,在6 种不同的界面中,B 终端的Ti/TiB2界面稳定性均优于Ti 终端的界面,且B 终端的孔穴位堆垛界面( BTH) 和Ti 终端的心位堆垛界面( TTC) 分别是两种终端最稳定的界面.
陶瓷颗粒增强金属基复合材料界面结合性能研究目前是国内外学者研究热点,但对于SiC/Mg界面结合性能研究未见报道.基于密度泛函理论的第一性原理对于界面特性研究有着优势,可以从微观方面研究作用机理和界面特性[10,12-15],可以满足对SiC/Mg 金属-陶瓷复合材料界面结合机制的研究.本文通过第一性原理研究了4 种搭建结构的3C-SiC(111) /Mg(0001) 界面模型,通过分析理想粘附功和电子结构得出稳定的结构.
2 计算方法及模型
2.1 计算步骤和细节
本文所有的计算都在MATERIALS STUDIO 软件的CASTEP 模块下进行[16,17],描述电子与电子之间作用的交换关联势泛函的选取为GGA-PBE,优化算法选取BFGS,赝势采用平面波超软赝势.能量的计算采用自洽迭代方法( SCF) ,自洽迭代的收敛阈值为1.0 ×10-6eV /原子,能量的截断点为380 eV,能量计算允许的最大自洽迭代次数为100,几何优化时作用在原子上的最大力应不超过0.03 eV/Å,最大应力应不超过0.05 GPa,最大位移应不超过0.001 Å,第一布里渊区K 值为6 ×6 ×1.
Mg 原胞由两个Mg 原子构成,空间群为P63/mmc( No.194) ,晶格参数为a=b=3.2094 Å,c=5.2105 Å,α=β=90°,γ=120°.3C-SiC 又名β-SiC,具有闪锌矿结构,空间群为F-43m,它的原胞由4 个Si 原子和4 个C 原子组成,四个相近的原子将中间的异种原子包围而形成一个正四面体.晶格参数a=4.348 Å.
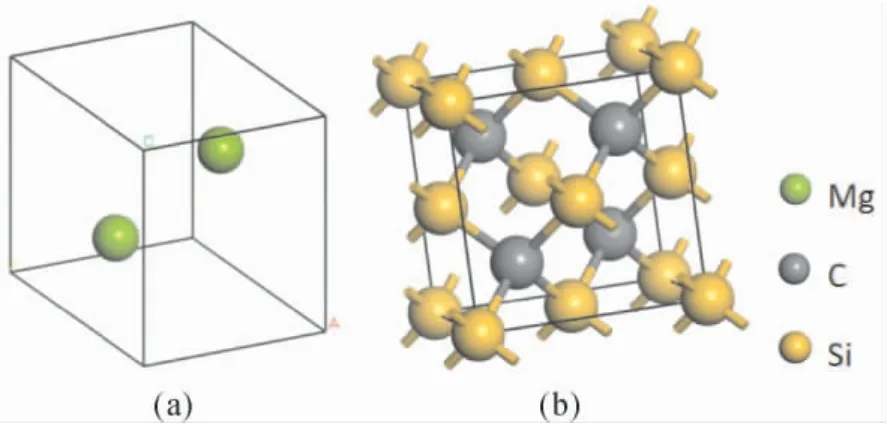
图1 ( a) Mg 的晶体结构; ( b)3C-SiC 的晶体结构Fig.1 ( a) Crystal structure of Mg; ( b) Crystal structure of 3C-SiC
2.2 模型建立
图2 显示的是3C -SiC(111) /Mg(0001) 的界面模型,通过在10 层的3C -SiC( 111) 上堆叠5层Mg(0001) 来实现,在Mg(0001) 侧的上方建立15 Å 厚度的真空层,防止表面原子之间的相互作用.对于3C-SiC(111) 是极性表面,有着两种封端结构,因此采用两种封端来模拟3C - SiC(111) /Mg( 0001) 的界面.本文还考虑了Mg(0001) 面和3C -SiC( 111) 面可以构建中心型和顶位型两种堆垛结构,总共建立4 个模型用于接下来的计算分析.
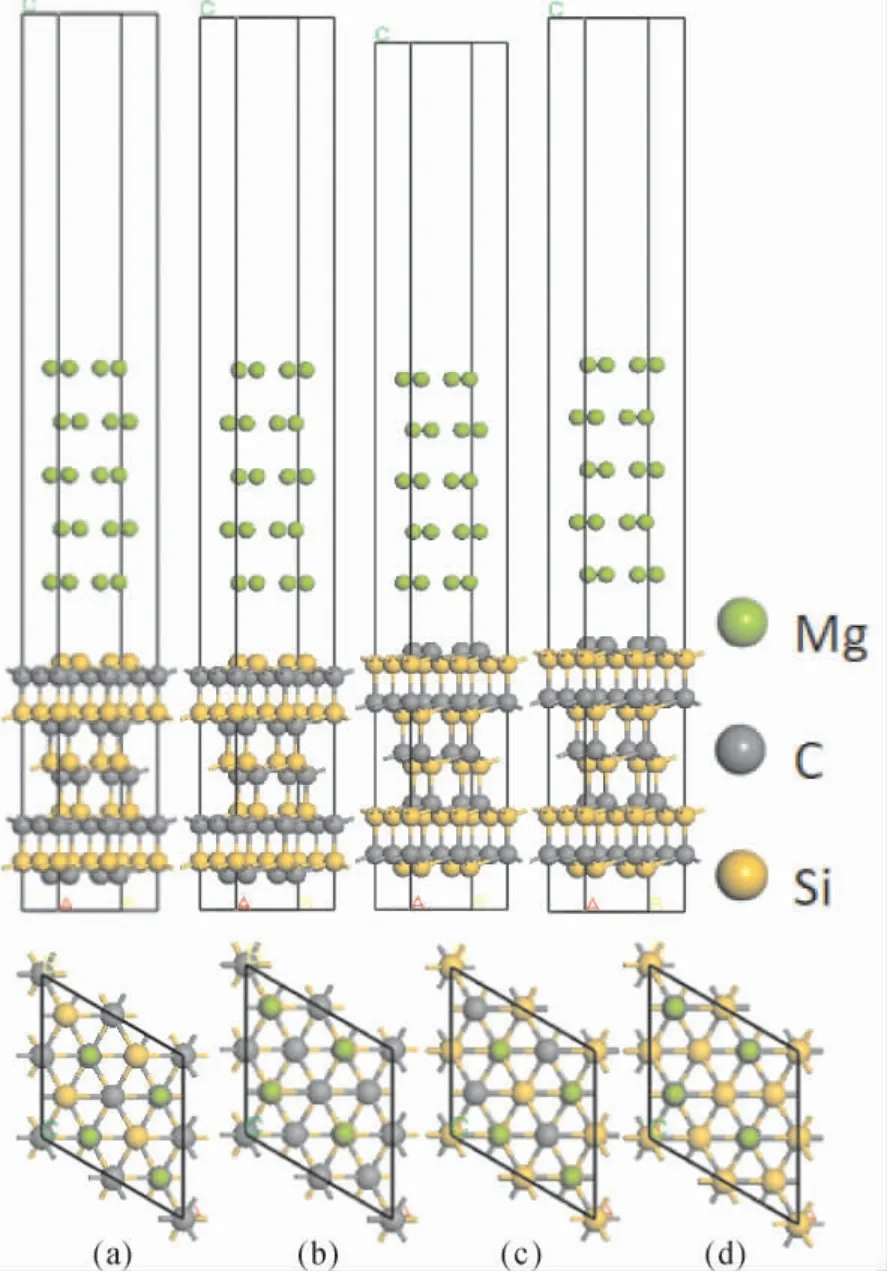
图2 3C-SiC(111) /Mg( 0001) Si 终端和C 终端的四种结构: ( a) Si 终端中心型; ( b) Si 终端顶位型; ( c) C 终端中心型; ( d) C 终端顶位型Fig.2 Four structures of Si-terminated and C-terminated 3C-SiC(111) /Mg(0001) : ( a) Center-site Si-terminated; ( b) Top -site Si -terminated; ( c) Center - site C - terminated;( d) Top-site C-terminated
3 结果与讨论
3.1 结构优化
结构优化计算过后,3C - SiC ( 111) /Mg(0001) 的4 种界面模型在z 轴方向原子都发生了位移,使得模型的界面间距产生了不同程度的缩小,达到了每个界面较稳定的状态.
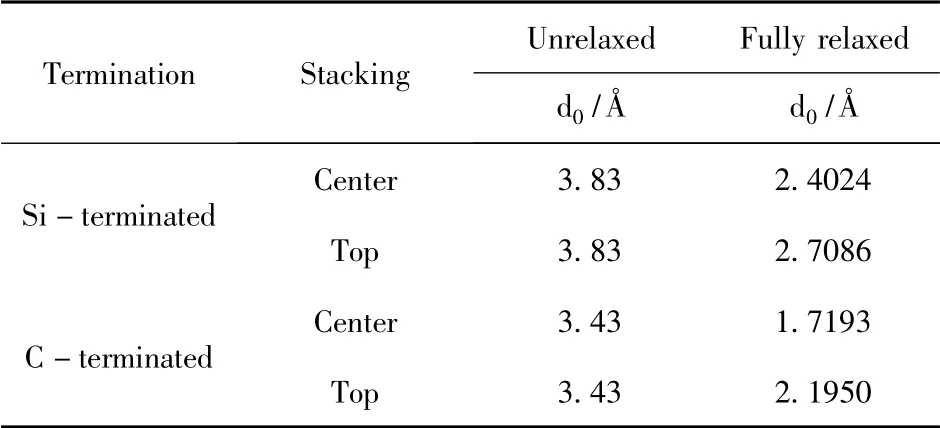
表1 3C-SiC(111) /Mg(0001) 界面结构优化前后的界面间距Table 1 Interface spacings before and after structure optimization of 3C-SiC(111) /Mg(0001)
3.2 理想粘附功
金属/陶瓷界面结合强度的表示通常用分离功这个物理量,分离功的对立是粘附功,粘附功的含义是指两个自由表面结合形成一个界面所释放的能量,其计算公式如下[18]:
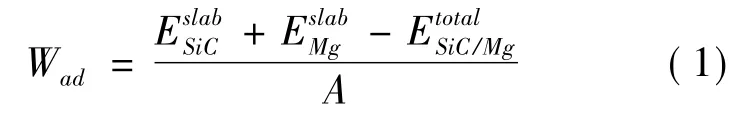
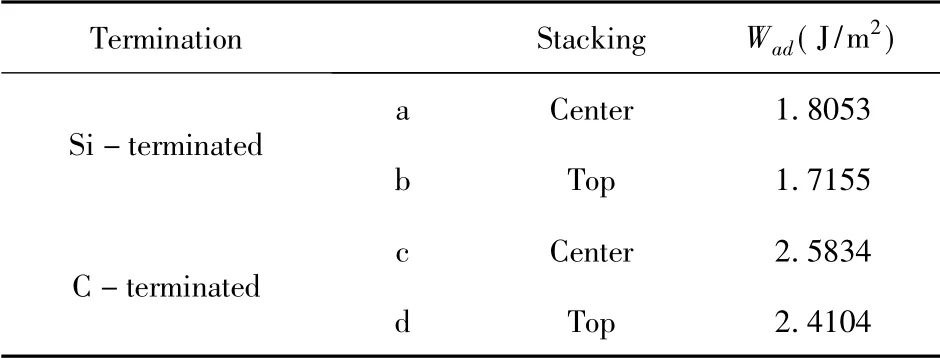
表2 3C-SiC(111) /Mg(0001) 界面的粘附功Table 2 Adhesion work of 3C - SiC( 111) /Mg( 0001)interface
结构优化之后,4 个模型在z 轴方向都发生了界面距离不同程度的减少,界面间距与界面结合强度有着紧密的关系,而在4 个3C - SiC(111) /Mg(0001) 界面模型中,C 封端模型的界面间距比Si 封端模型更小,粘附功而言,C 封端模型的更大,相对来说,C 封端模型明显更稳定.该结果与以往的研究[19-21],当SiC 与金属结合时,用C 封端与金属面接触往往表现出更强的稳定性相一致.而在Si 封端和C 封端的模型中,各自的中心型模型都优于顶位型模型,有着更小的界面间距(2.4024 Å 和1.7193 Å) 和更大的粘附功(1.8053 J/m2和2.5834 J/m2).
3.3 电子结构分析
图3( a) 和图3 ( b) 分别是3C -SiC(111) /Mg(0001) Si 终端中心型结构的电荷密度分布和电荷密度差,图3( c) 和图3 ( d) 分别是3C - SiC(111) /Mg(0001) C 终端中心型结构的电荷密度分布和电荷密度差.图3( a) 和图3 ( c) 中Mg 侧和SiC 侧中间有电荷积累.SiC 侧的电荷密度主要聚集在C 原子附近,这是因为C 原子相对于Si 原子有较强的电负性.图3( b) 和图3( d) 可以看出,Si 原子附近主要是代表着失去电子的蓝色区域,C 原子附近主要是代表得到电子的红色区域.在界面处存在电子云重叠,图3( d) 的电子云重叠现象明显于图3( b).界面处的Mg 原子失去的电子进入界面,与界面处的Si 原子和C 原子的电荷形成离子键/共价键.图3( d) 在界面处积聚电子的现象比图3( b) 更显著,表明C 端结构比Si 端结构的界面结合更稳定.这个结论与粘附功的分析结果相一致.
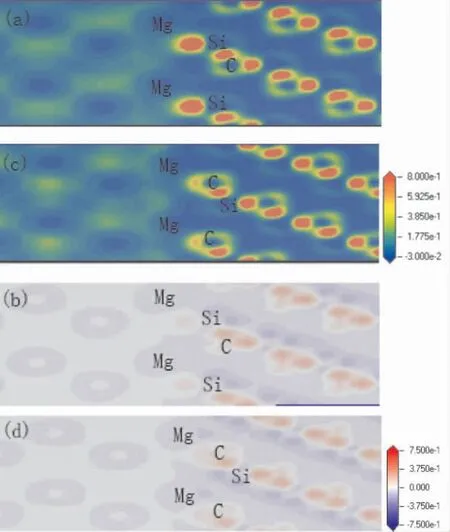
图3 中心型电荷密度分布图和差分电荷密度图:( a) Si 终端电荷密度分布图; ( b) Si 终端差分电荷密度图; ( c) C 终端电荷密度分布图;( d) C 终端差分电荷密度图Fig.3 Charge density distributions and charge density differences of center -site: ( a) charge density distribution of Si - terminated; ( b) charge density difference of Si - terminated; ( c)charge density distribution of Si - terminated;( d) charge density difference of Si-terminated
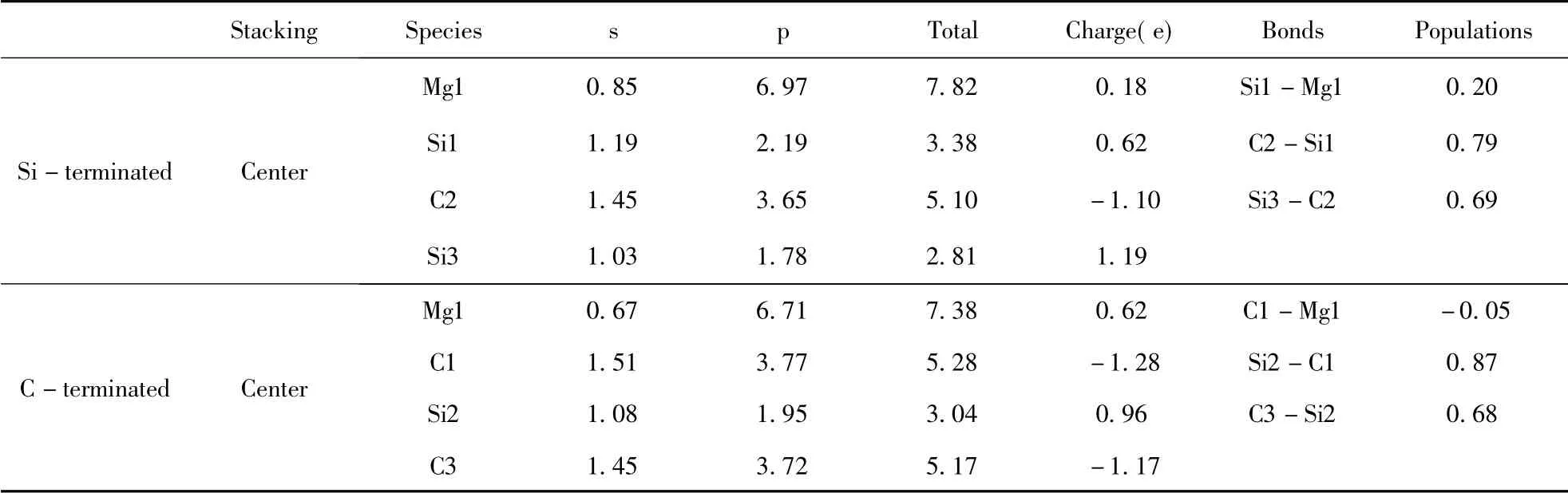
表3 中心型两种终端结构界面原子的Mulliken 电荷Table 3 Mulliken charges of atoms at center-site
布居数可以反应出电子在界面处的散布状况以及衡量成键类型和强弱,键布居数越靠近0 代表离子性越强,越靠近1 代表共价性越强.在Si端3C -SiC( 111) /Mg( 0001) 界面上,界面的Mg原子失去部分电子变成+0.18 价,与界面上的Si原子的重叠布居数为0.20,处于键合状态.与Si端相比,C 端3C -SiC(111) /Mg(0001) 界面上的Mg 原子失去的电子( +0.62) 更多,与界面上的C原子成弱反键状态,重叠布居数为-0.05.Si 端Mg 原子与相邻原子形成的Si -Mg 键的布居数大于C 端Mg 原子与相邻原子形成的C -Mg 键,Si-Mg 键的离子性弱于C -Mg 键.从表中可以比较得出,Si 端与C 端3C-SiC(111) /Mg(0001) 界面处的Si-C 键的重叠布居数(0.79 和0.87) 大于内部的Si-C 键的重叠布居数(0.69 和0.68) ,表明界面改变了Si-C 的共价键性质.
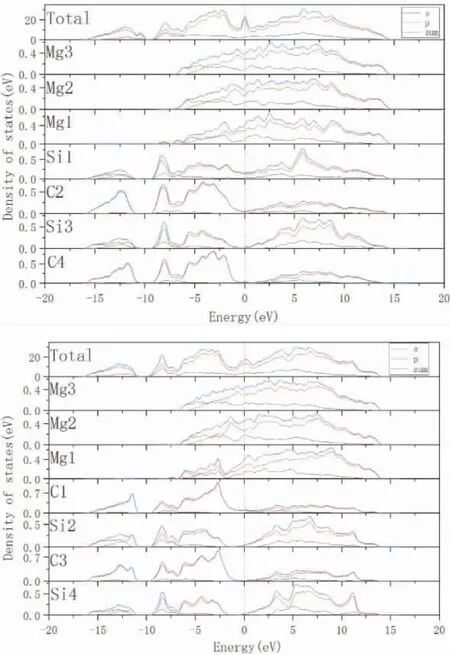
图4 3C-SiC(111) /Mg(0001) 界面态密度图: ( a)Si 终端中心型结构; ( b) C 终端中心型结构Fig.4 Density of states of 3C -SiC( 111) /Mg( 0001)interface: ( a) Center - site Si - terminated;( b) Center-site C-terminated
从态密度的计算结果可以看出界面处原子的成键性质,离界面比较远的原子对界面的影响很小,因此计算了3C-SiC(111) /Mg(0001) 中心型结构两种终端( C 终端和Si 终端) 的界面部分原子的态密度.在图5( a) 中,Mg 侧界面Mg 原子的态密度曲线不同于内部的Mg 原子的态密度曲线,Si侧原子的态密度曲线也出现不同.计算结果显示,Si 侧界面Si 原子的态密度曲线高度低于内部C 原子的态密度曲线高度( 从-15.9 eV 到-10.1 eV) ,表明SiC 侧界面Si 原子的电子发生转移到了Mg侧的界面Mg 原子,从而形成了离子键.由于Sisp、C-sp、Mg-sp 在-9.5 eV 到14.6 eV 之间的轨道杂化,表明有共价键的存在.在费米能级附近,Mg 原子和Si 原子的明显的占据态表明有金属键产生.C 终端原子态密度的分析和Si 终端原子态密度的分析相近,Si 终端和C 终端界面原子成键类型主要是共价键、离子键和少量金属键.
4 结 论
本文采用第一性原理的方法计算了3C -SiC(111) /Mg(0001) 界面的粘附功和电子结构,考虑了界面的中心型和顶位型两种堆积结构和Si 终端和C 终端两种端接结构.计算后可以得出以下结论:
(1) 结构优化后,除了界面间距均有或多或少的减少外,这4 种界面模型的结构没有发生特别大的变化.
(2) 中心型结构的粘附功大于顶位结构,C终端结构的粘附功大于Si 终端结构,C 终端中心型的结构是四个结构中最稳定的.
(3) 根据电荷密度分布图、差分电荷密度图、Mulliken 电荷和态密度图分析,这两种界面处存在离子键、共价键和少量金属键的混合.
猜你喜欢
教育家(2022年19期)2022-05-13
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年10期)2021-11-22
成都信息工程大学学报(2021年2期)2021-07-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
华东师范大学学报(自然科学版)(2020年1期)2020-03-16
燕山大学学报(2015年4期)2015-12-25
新高考·高一物理(2015年6期)2015-09-28
