
B对Mo等元素在奥氏体钢/Cr2O3界面占位倾向影响的理论研究
2022-03-04 02:33韩培德
原子与分子物理学报 2022年4期
闫 新,董 楠,张 翊,韩培德
( 太原理工大学 材料科学与工程学院,太原 030024)
1 引 言
超级奥氏体不锈钢中因含有更高的铬、镍、钼、氮等元素而具有更加优异的抗腐蚀性能,被广泛应用于烟气脱硫、海水淡化、纸浆漂白、垃圾焚烧等领域,尤其适用于海水、低速冲刷等严苛的腐蚀环境[1-3].然而,超奥不锈钢是不锈钢中生产工艺极其复杂、加工难度极大的钢种之一,较高的钼含量极易导致其在凝固、热加工过程中产生偏析,出现开裂等破坏,同时过多的析出相也严重降低了其耐蚀性能[4,5].
超级奥氏体不锈钢耐蚀性能主要取决于致密的Cr2O3,大量Mo 等合金元素的加入可促进钝化膜的致密性,Mo 通常认为以形成具有缓释作用的钼酸盐来发挥作用[6,7],具有极高的耐点腐蚀和耐缝隙腐蚀性能.Dou 等[8]分析了S31254 在模拟烟气脱硫气氛中钝化膜的组成和结构,发现Mo以MoO3-的形式分布于次表层发挥耐蚀作用.近期的研究发现,在S31254 中引入微量的B,可以抑制析出相的析出,同时耐蚀性也得到了提高,耐蚀性提高的主要原因是由于含硼S31254 钝化层中Cr、Mo 含量明显增加[9,10].现有的实验表征手段,对Fe/Cr2O3界面结构、钝化层进行了成分、结构表征,获得了元素分布信息.但是原子层次解释奥氏体不锈钢钝化过程中Mo 等合金元素在不锈钢基体、氧化层以及界面中的迁移、扩散机制,尤其超奥钢中B 对这些元素占位的影响仍不清楚.奥氏体不锈钢中除Mo、Cr、Ni 外,Mn、Si 也均为常加元素.Mn 常常优先于Cr2O3生成尖晶石氧化物,Ni 会减缓阳离子在Cr2O3氧化膜中的扩散,并阻止FeCr2O4+ Fe2O3的形成,从而增强合金的抗氧化性能.第一性原理计算方法已广泛应用于研究材料原子层次的结构和相关性能,在材料微观结构模拟、原子、电子结构分析等方面均发挥出极大的作用.Li 等[11]采用第一性原理研究了B 对Mo 在Fe-Cr -Ni∑5(210) 晶界偏析行为的影响.Si 对Cr2O3氧化层的形成有一定影响,在富Cr 氧化层下生成的SiO2能阻止Cr 离子向外扩散[12].Heo 等人[13]研究了P 和B 对Fe 基合金、Ni3Al 及Ni 金属晶界的影响,Mn 对Fe -12Mn 二元合金晶界的影响,以及B、C、P 在α-Fe 晶界结合稳定性及对其强韧性的影响.但相关B、Mo 等在Fe/Cr2O3钝化层界面占位倾向的研究,及对Mo 等在fcc -Fe/Cr2O3界面偏析行为尚缺乏深入理解.本文拟构建fcc -Fe/Cr2O3界面模型,采用第一性原理计算方法,分析Mo 等合金元素在体系中的偏析倾向,以及B 对Mo 等合金元素分布行为的影响,为提高Mo 等合金元素不锈钢的耐蚀性能提供理论指导.
2 计算方法和模型建立
2.1 计算方法
采用VASP[14,15]软件包完成.交换关联函数均采用广义梯度近似GGA[16]中的PBE[17]泛函,原子核和价电子间的相互作用采用PAW 方法处理[18].平面波的截断能为400 eV,布里渊区K 点取样[5 ×5 ×1],迭代计算时自洽场的收敛精度为10-6eV/atom,每个原子上的力收敛标准为0.01 eV/Å.此外Fe、Cr、O、B、Mo、Mn、Si、Ni 的价电子构型分别为3d64s2、3d54s1、2s22p4、2s22p1、4d55s1、3d54s2、3s23p2、3d84s2.
2.2 模型构建
超级奥氏体不锈钢为面心立方结构[19],选取fcc-Fe 作为基体结构,并与Cr2O3[20]进行界面模型的搭建,构建的fcc -Fe(111) /Cr2O3(0001) 界面模型,如图1 所示.首先计算fcc -Fe 单胞结构参数为3.44 Å,与先前计算的3.45 Å[21]结果相符.Cr2O3晶体在室温下属于六方晶系的R-3C空间群,其中O2-按照六方紧密堆积排列,Cr3+占据氧原子间2/3 的八面体间隙位置,其余1/3的八面体间隙空置,计算的Cr2O3单胞结构参数为a=b=5.075,c=13.815 Å,α=β=90°,γ=120°,与实验值[22]非常接近.模型fcc -Fe(111)/Cr2O3(0001) 包含两种终端结构: O 终端和Cr 终端.结构优化后对比,O 终端界面结构( -246.560 eV) 比Cr 终端稳定( -246.273 eV) ,该计算结果与文献报道一致[23,24],下述将选择O 终端结构模型进行进一步的研究.
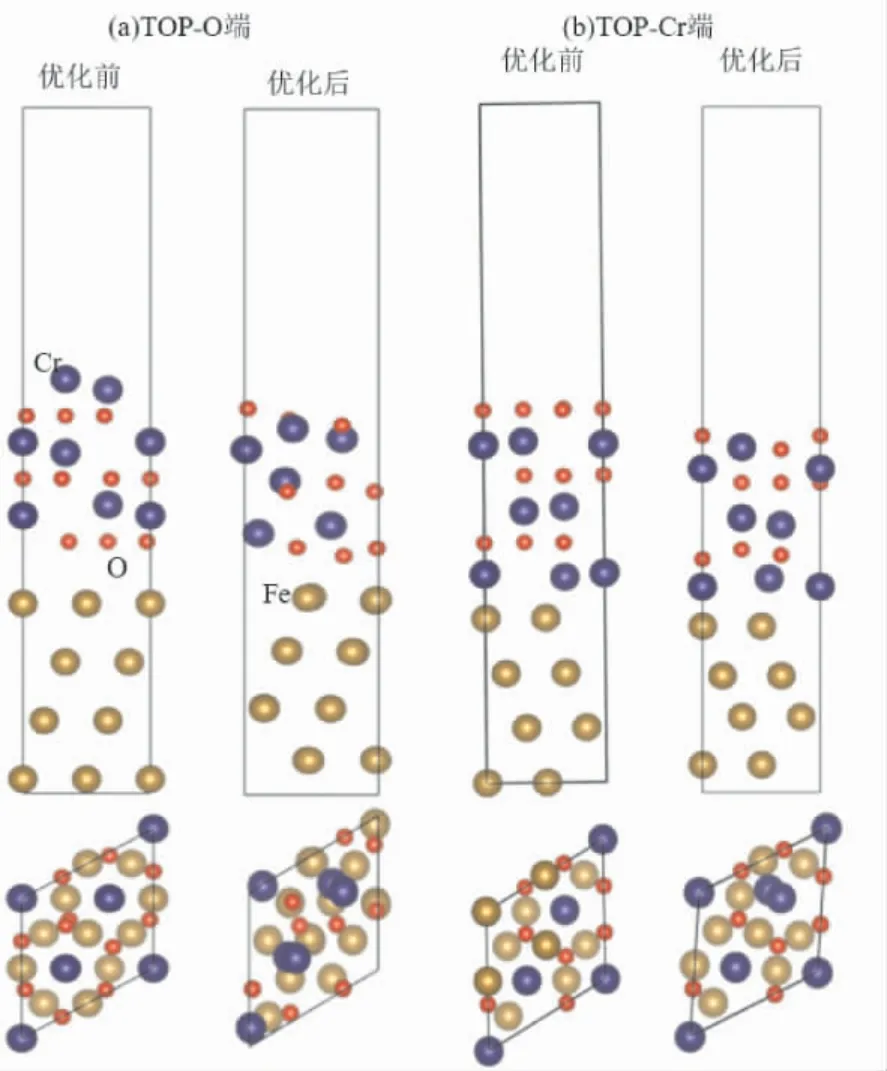
图1 fcc - Fe( 111) /Cr2O3( 0001) 界面结构,( a)Top-O 端和( b) Top-Cr 端优化前后的模型结构Fig.1 Optimized structures of ( a) Top -O and ( b)Top-Cr termination in fcc -Fe( 111) /Cr2O3(0001)
(1) 结合能[25]
在热力学和动力学范畴内,能量是判断系统稳定性的基本参量.它不仅能反映物质的宏观性能,还能反映微观粒子的相互作用.体系的结合能数值判断体系结构的稳定性能,结合能数值小于零,体系能稳定存在.结合能计算公式(1) 所示.
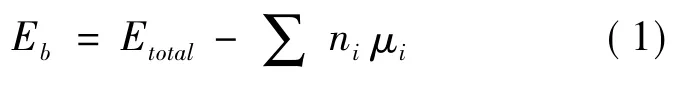
其中,Eb为结合能,Etotal表示晶胞的总能,μi为孤立态时各表面或晶界中单个原子的能量,ni为体系中各元素的原子总数.
(2) 偏聚能[24,25]
合金元素在界面处的偏析趋势,可通过计算界面偏聚能Eseg来衡量,本文将计算Mo、Mn、Ni、Si、B 原子在界面的偏聚行为及对界面的影响,偏聚能计算公式(2) 所示.
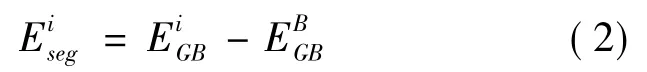
3 结果与讨论
3.1 合金元素在fcc -Fe(111)/Cr2O3(0001)界面体系稳定性及偏析倾向
3.1.1 界面体系稳定性
图2 为合金元素Mo、Ni、Mn、Si 在界面体系中的结合能.结合能均小于零,说明以上4 种原子固溶于界面体系构成的结构可稳定存在.Mn原子固溶于体系的结合能更负,说明Mn 的固溶有利于提高界面体系的结构稳定性.

图2 Mo、Mn、Ni、Si 固溶于fcc -Fe(111) / Cr2O3(0001) 后体系的结合能Fig.2 The values Eb of Mo,Mn,Ni,Si solid solution in fcc-Fe(111) / Cr2O3(0001)
3.1.2 合金元素界面偏析倾向
图3a 为 Mo、Mn、Ni、Si 置 换 fcc - Fe(111) /Cr2O3(0001) 界面体系中不同层面的Fe 原子的模型图; 图3b 为Mo、Mn、Ni、Si 合金原子位于体系不同位置对应的偏析能.原子偏聚能可以反映其是否向晶界位置偏聚及偏聚的可能性.偏聚能为负,代表合金元素向晶界位置偏聚,同时绝对值的数值越大,表示原子越易向晶界位置偏聚.偏聚能为正值,代表合金元素不易偏聚,易均匀分布于基体.
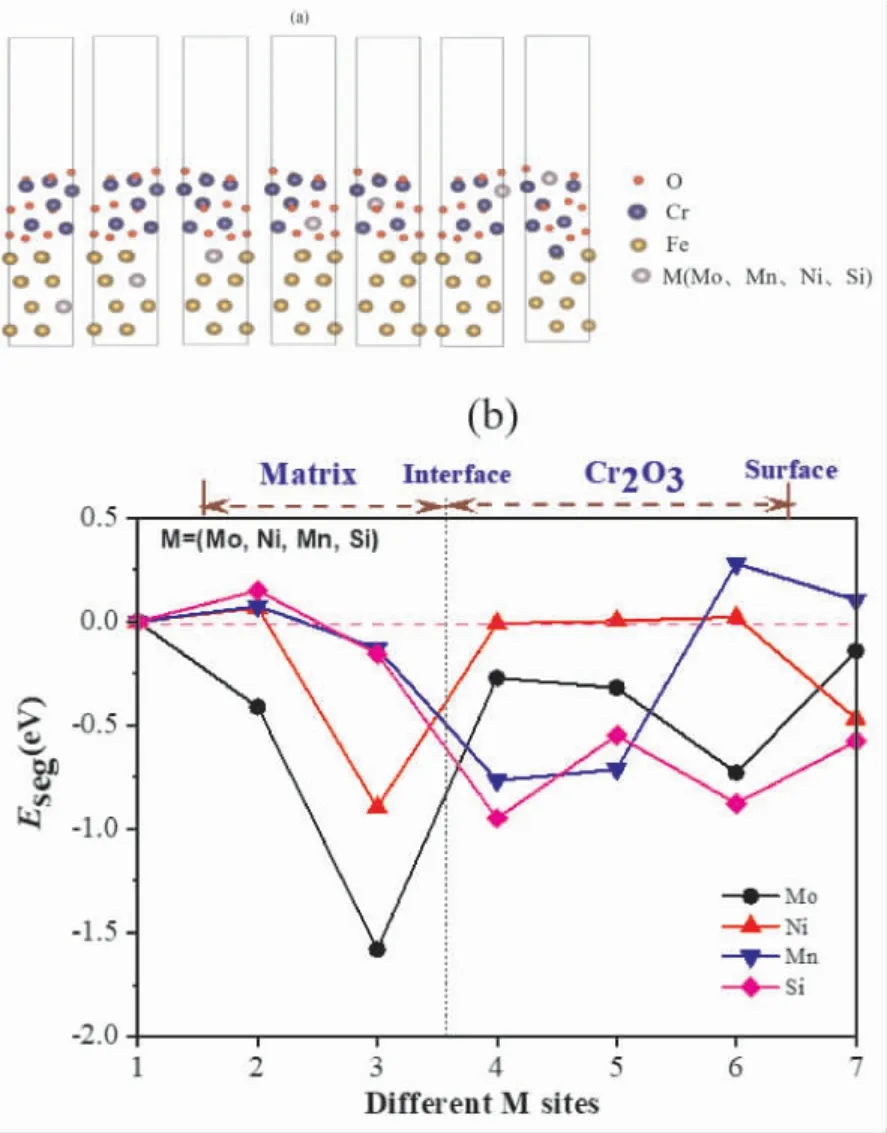
图3 Mo、Mn、Ni、Si 在fcc - Fe( 111) /Cr2 O3(0001) 中占位( a) 及其偏聚能( b)Fig.3 The different diffusion sites of Mo atoms at the fcc - Fe( 111) /Cr2O3( 0001) ( a) and their segregation energies ( b)
从偏聚能Eseg可以看出,Mo、Ni 更倾向于分布于界面基体侧,Mo 有由界面基体侧向Cr2O3氧化层扩散的趋势,Mo 较Ni 更易于扩散到界面氧化层中; Mn、Si 更倾向于分布界面体系的氧化层中; 且Mo 更易偏聚于界面基体侧,Mn、Si 更倾向由基体界面基体侧向Cr2O3氧化层中扩散,且Si氧化层中的偏聚倾向大于Mn.结合S32654 高温氧化实验结果来看[23],氧化开始阶段,氧化层表面先形成的氧化物中Mn 含量很高,表明Mn 在高温氧化过程中易由基体向氧化层扩散; 从S32654高温氧化后氧化层中各元素分布来看,Mo 主要集中在基体和氧化层( Cr2O3为主) 之间,并且氧化层中也观察到了Mo 的存在,但是过高的氧化温度下Mo 会发生烧损.表明本计算中Mo、Mn 易在界面基体侧及氧化层的结果和实验现象相吻合.
3.2 B 在fcc -Fe(111)/Cr2O3(0001)界面体系的偏析倾向
图4 结构模型为间隙原子B 在fcc - Fe(111) /Cr2O3( 0001) 界面体系中不同层面所处位置,B 作为间隙原子固溶在fcc -Fe( 111) /Cr2O3(0001) 界面体系中.其中,位点2 代表B 处于基体Fe 中,位点3 处于界面基体Fe 侧,位点4 处于相界面处,位点5 处于Fe(111) 和Cr2O3(0001)相界面Cr2O3( 0001) 侧,位点6、7 处于氧化膜Cr2O3(0001) 中.体系的结合能均小于零,说明B溶于界面体系具有更为稳定的构型.相比之下,B 处于界面的基体侧和氧化物侧更稳定.图4 所示.界面处的B 原子偏聚能数值大于零,表明B易偏析于相界面基体侧( 位点3).
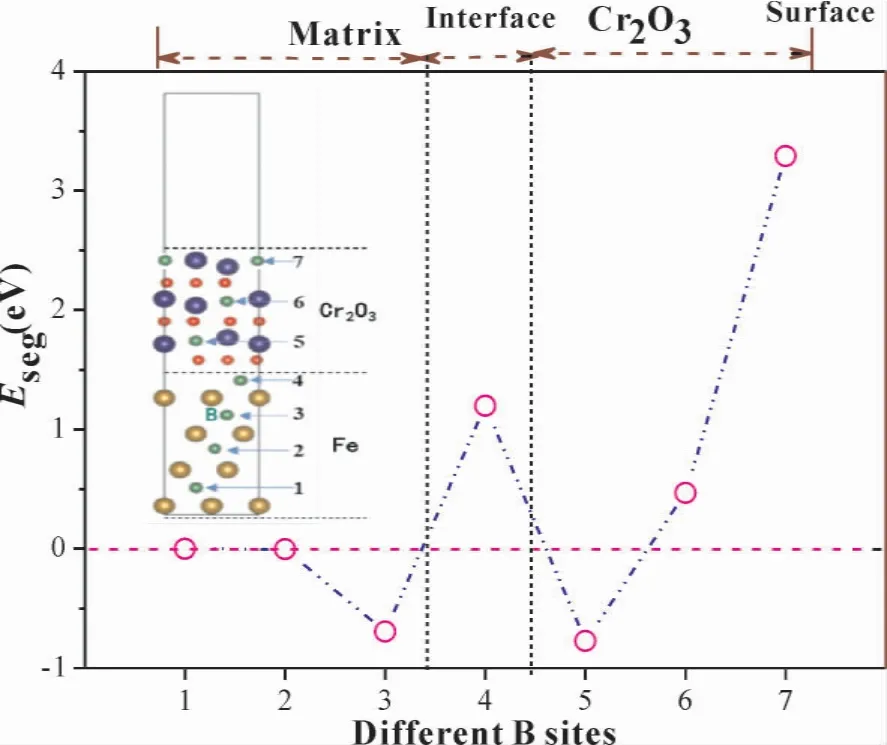
图4 B 在fcc-Fe( 111) / Cr2O3( 0001) 界面不同位置分布结构模型及偏聚能Fig.4 Structure models and segregation energies of B atom at different diffusion sites of fcc - Fe(111) / Cr2O3(0001) interface
Mo、B 倾向分布于界面基体侧,且B 较Mo的偏析倾向更大,优先分布于相界侧,很有必要进一步分析B 处于基体侧时,对Mo、Mn、Ni、Si原子在fcc-Fe(111) /Cr2O3(0001) 中的占位倾向的影响.
3.3 B 对合金元素在fcc - Fe(111)/Cr2 O3(0001)界面体系稳定性及偏析倾向的影响
3.3.1 B 对稳定性的影响
Mo、Mn、Ni、Si 原子固溶于有B 的fcc -Fe(111) /Cr2O3( 0001) 体系的结合能均小于零( 图5) ,说明以上4 种原子固溶于该体系所形成的结构均能稳定存在,具体来看Mo、Mn 固溶于界面体系的结合能均低于含硼fcc - Fe( 111) /Cr2O3(0001) 界面体系,说明Mo、Mn 的固溶有利于提高界面体系的结构稳定性.
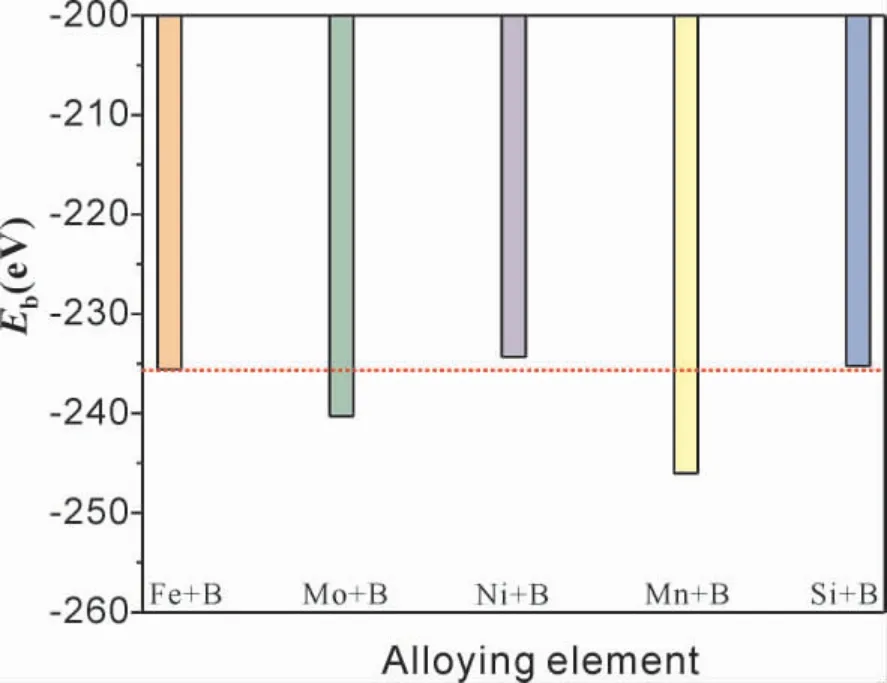
图5 Mo、Mn、Ni、Si 固溶于含B 的fcc - Fe(111) / Cr2O3(0001) 后体系的结合能Fig.5 The values Eb of Mo,Mn,Ni,Si solid solution in fcc-Fe(111) / Cr2O3(0001) with B atom
3.3.2 B 对合金元素偏析倾向的影响
结合图4 计算结果,B 在fcc -Fe( 111) /Cr2O3(0001) 体系中位点3 处稳定( 图4) ,故分析其对Mo、Mn、Ni、Si 在fcc -Fe( 111) 中占位倾向的影响,重点分析fcc -Fe( 111) 和Cr2O3( 0001)的界面侧以及氧化膜Cr2O3(0001) 中位置处情况.当Mo、Mn、Ni、Si 等分布于氧化层时,在基体中留下的空位由Cr 原子填充.图6 结构模型表示了Mo、Mn、Ni、Si 置换原子在含硼fcc - Fe(111) /Cr2O3( 0001) 界面体系中不同层面所处位置,依据公式(2) ,计算各模型所对应的偏聚能,如图6 所示.B 处于界面基体侧时,B 可延缓Mo向界面基体侧的偏聚,且不易分布于氧化层中;B 可进一步促进Mn 由基体向界面体系基体侧的偏聚; 同时B 使得Ni 更易于在界面体系中趋于均匀分布.这一结果,可以看出B 的作用,可以缓解S32654 高温氧化过程[23]中Mo 的扩散、烧损、挥发问题,B 的引入延缓了Mo 由基体向界面基体侧的偏聚,同时又能阻止Mo 由界面向氧化层的扩散.
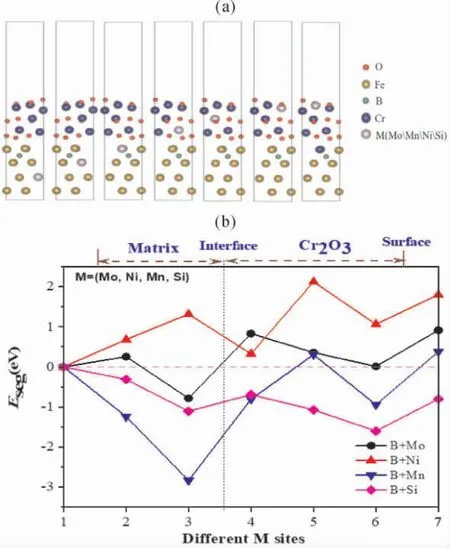
图6 Mo、Mn、Ni、Si 在含硼fcc-Fe(111) / Cr2O3(0001) 中占位( a) 及其偏聚能( b)Fig.6 The different diffusion sites of Mo,Mn,Ni,Si atoms at the fcc-Fe(111) / Cr2O3(0001) with B atom ( a) and their segregation energies ( b)
3.4 电子性质分析
图7( a-d) 为间隙原子B 位于fcc-Fe(111) /Cr2O3( 0001) 界面体系基体侧位点3,置换原子Mo、Mn、Ni、Si 位于fcc-Fe(111) /Cr2O3(0001)界面体系基体侧,Mo +B、Mn +B、Ni +B、Si +B 共同溶于fcc -Fe( 111) /Cr2O3( 0001) 界面体系的态密度图.图7( a) 所示,fcc -Fe( 111) /Cr2O3(0001) 在费米能级处的能量为45.47 states/eV,体系中溶入一个Mo 原子后,费米能级总能量降为42.11 states/eV; 体系中溶入一个B 原子后,费米能级处总能量增加到49.11 states/eV; Mo 和B 复合溶入体系后,费米能级处的能量为42.34 states/eV,与fcc -Fe( 111) /Cr2O3( 0001) 能级相近.具体由图中分态密度来看,费米能级处Mo对费米能级处的贡献大于B 的贡献,另外远离费米能处,位置约-21.44 ~-18.21 eV 区域、 -8.4 ~2 eV 区域,也出现了大小不同的峰,从峰值高低看,由于仅有一个原子参与峰值较小,B对能级处的贡献大于Mo 的贡献.具体来看, -21.44 eV ~-18.21 eV 区域的峰,主要由B 原子2s 轨道形成,还有O 的2s 轨道、Cr 的3d 轨道和Mo 的4d 轨道( Mo 处于界面基体侧) 上的电子贡献; -8.36 eV ~2 eV 区域则主要是由Fe、Cr 和Mo 原子3d 轨道的电子、O 的2p 轨道电子以及B的2s、2p 轨道电子贡献的.说明B 的引入加强了其与邻近原子的相互作用,尤其B、Mo 复合溶入fcc-Fe( 111) /Cr2O3( 0001) 界面体系有利于提高体系的电化学稳定性.
图7( b) 为Mn +B 溶于fcc -Fe( 111) /Cr2O3(0001) 界面体系的态密度图.Mn 和B 复合溶入体系后,费米能级处的能量为44.65 states/eV.具体由图中分态密度来看,费米能级处Mn 对费米能级处的贡献远大于B 的贡献,另外远离费米能处,位置约-21.09 ~-17.59 eV 区域,出现了大小不同的峰,从峰值高低看,B 对能级处的贡献和Mn 相近.具体来看,-21.09 ~-17.59 eV区域的峰,主要由O 的2s 轨道、还有B 原子2s轨道、Cr 的3d 轨道和Mn 的3d 轨道上的电子贡献; -8.04 ~2 eV 区域则主要是由Fe、Cr 和Mn原子3d 轨道的电子、O 的2p 轨道电子以及B 的2s、2p 轨道电子贡献的.说明B、Mn 的引入加强了它们与邻近原子的相互作用.具体到实际的奥氏体不锈钢其是多元素的组合,其影响远比单一元素的作用复杂.
图7( c) 为Ni + B 溶于fcc - Fe( 111) /Cr2O3(0001) 界面体系的态密度图.Ni 和B 复合溶入体系后,费米能级处的能量为43.99 states/eV.由图中分态密度,费米能级处Ni 对费米能级处的贡献远大于B 的贡献,远离费米能处,位置约-21.70 eV ~-18.34 eV 区域,出现了高低不同的峰,从峰值高低看,B 对能级处的贡献和Ni 相近.具体来看, -21.70 eV ~-18.34 eV 区域的峰,主要由O 的2s 轨道、B 原子2s 轨道、Cr 的3d 轨道和Ni 的3d 轨道上的电子贡献; -10.28 eV ~2.44 eV 区域主要是由Fe、Cr 和Ni 原子3d轨道的电子、O 的2p 轨道电子以及B 的2s、2p轨道电子贡献的.说明B、Ni 的引入加强了它们与邻近原子的相互作用.
图7( d) 为Si + B 溶于fcc - Fe( 111) /Cr2O3(0001) 界面体系的态密度图.Si 和B 复合溶入体系后,费米能级处的能量为42.30 states/eV,与fcc-Fe( 111) /Cr2O3( 0001) 相比原子数减少.具体由分态密度来看,费米能级处Si 中电子对费米能级处的贡献大于B 的贡献,另外远离费米能处,位置约-21.37 eV ~-17.57 eV 和-11.6 eV~-9.47 eV 区域,出现了大小不同的峰,从峰值高低看,B 对能级处的贡献和Si 相近.-21.37 eV~-17.57 eV 区域的峰,主要由O 的2s 轨道、B原子2s 轨道、Cr 的3d 轨道和Si 的3p 轨道上的电子贡献; -9.25 eV ~3.00 eV 则主要是由Fe 和Cr的3d 轨道电子、Si 原子的3p 轨道、O 的2p 轨道电子以及B 的2s、2p 轨道电子贡献的.说明B、Si的引入加强了它们与邻近原子的相互作用.
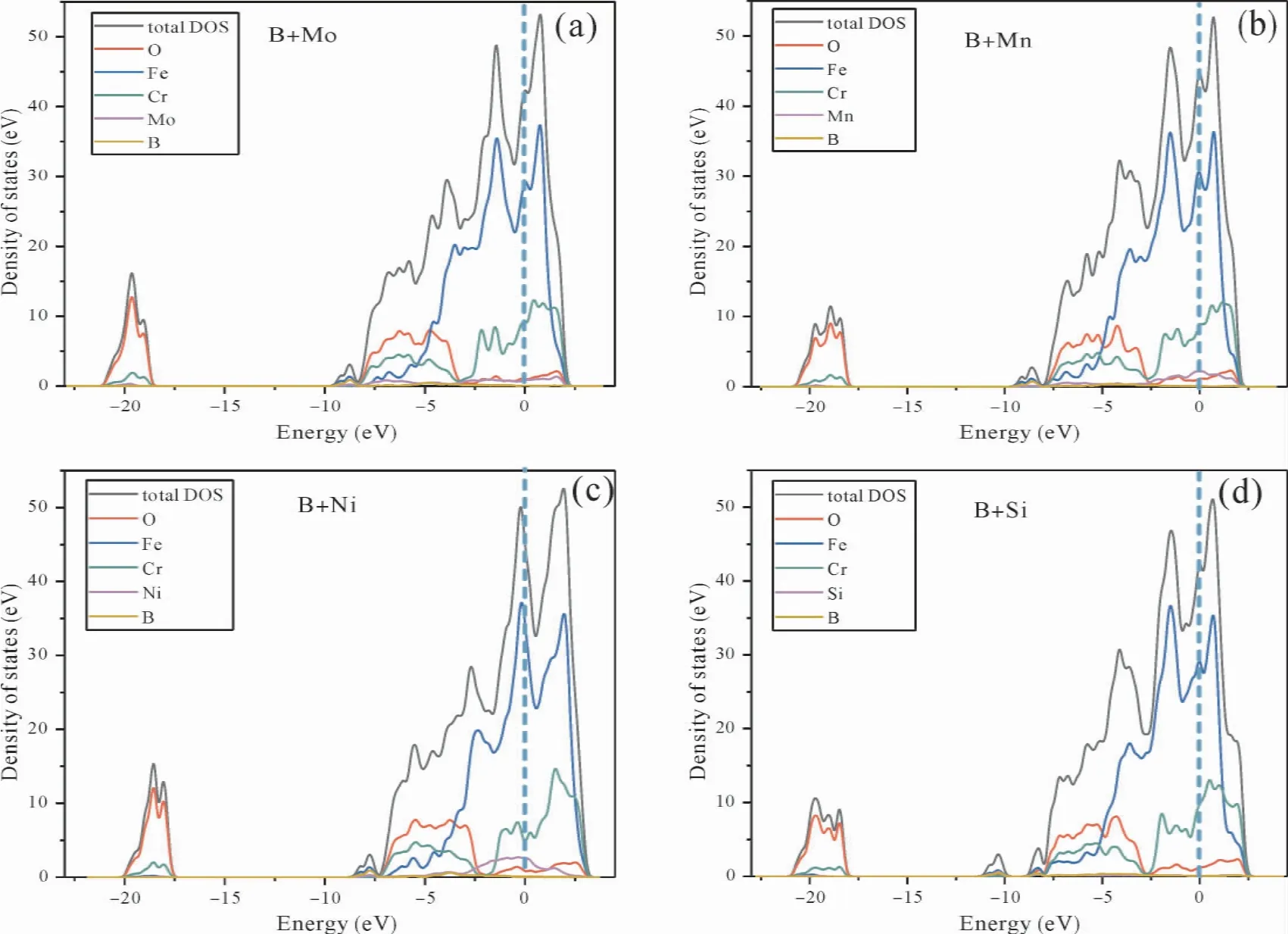
图7 Mo+B、Mn+B、Ni+B、Si+B 溶于fcc-Fe(111) /Cr2O3(0001) 界面体系的态密度图( a-d)Fig.7 Density of states ( DOSs) of fcc -Fe( 111) /Cr2O3( 0001) with Mo + B,Mn + B,Ni + B,Si + B( a-d)
4 结 论
结合超级奥氏体不锈钢成分,研究了Mo、Mn、Ni、Si、B 在fcc - Fe/Cr2O3界面占位倾向,并分析了B 对这些元素占位的影响.得出如下结论:
(1) Mo、Mn、Ni、Si、B 均可稳定存在于fcc-Fe/Cr2O3界面; Mo、Ni 更倾向分布于界面基体侧,Mn、Si 倾向分布于体系氧化层中; 处于晶界B,可抑制Mo 在界面基体侧的偏聚,促进Mn 向界面基体侧的偏聚,Ni 均匀分布于基体.
(2) Mo +B、Mn +B、Ni +B、Si +B 复合溶入fcc-Fe/Cr2O3界面体系后,费米能级处的能量分别为42.34 eV、44.65 eV、43.99 eV、42.30 eV,Mo、Mn、Ni、Si 中电子对费米能级处的贡献大于B 的贡献,远离费米能级处,O 的2s 轨道贡献最大,Mo +B、Mn +B、Ni +B、Si +B 复合溶入fcc-Fe(111) /Cr2O3(0001) 界面体系有利于提高界面体系的电化学稳定性.
猜你喜欢
物理学报(2022年8期)2022-04-27
中外文摘(2021年7期)2021-04-23
鞍钢技术(2021年2期)2021-04-20
华人时刊(2021年23期)2021-03-08
中华养生保健(2020年2期)2020-11-16
发明与创新·小学生(2020年10期)2020-10-19
河南冶金(2020年3期)2020-09-10
复旦学报(医学版)(2020年3期)2020-06-18
发明与创新·小学生(2019年12期)2019-12-05
数码设计(2017年4期)2017-11-01
