
RE2O3(RE=Lu,Y,Sc)的氧空位第一性原理计算研究
2022-03-04 02:32陈士杰郭海波
原子与分子物理学报 2022年4期
陈士杰,冯 鹤,郭海波
( 上海大学 材料科学与工程学院,宝山 200444)
1 引 言
稀土倍半氧化物( 化学式可写为RE2O3,其中RE 代表稀土元素) 是一类极具应用前景的氧化物型闪烁体材料.相比其他类型的闪烁体材料,它们具有极高的密度,极大的光输出.RE2O3具有立方对称性,结构稳定,禁带宽度约在4 -6 eV,是一种理想的光学材料.在稀土倍半氧化物中,Lu2O3、Y2O3、Sc2O3是常用的发光应用基体材料,可用于闪烁[1,2]、激光介质[3,4]、磷光体[5]和电介质[6]等方面.掺Eu、Ce 等元素后,可获得不错的发光强度和光学性能,能够和很多传感器相耦合,达到探测的作用.但是由于氧空位等缺陷所导致的“余辉效应”影响着发光效率和能量转换率,始终是一个头疼的问题.
当高能射线、高能粒子[7]等通过某些晶体时,因射线或粒子的激发,会发射出荧光脉冲( 如紫外或可见光) 的功能材料被称为闪烁晶体[8].闪烁晶体的基体材料和激活剂多为稀土成分,属于稀土发光材料的范畴[9].闪烁晶体可制成探测器,应用前景广阔,主要包括核医学[10]、安检、工业无伤探伤、油井勘测、空间物理、高能物理和非破坏性探测等诸多领域[11].闪烁晶体根据发光特点可以分为本征发光体和非本征发光体.前者无需引入杂质,其自身可发光,大多有高密度和快衰减的特性,因为依赖材料自身合适的禁带宽度形成跃迁发光,但本征禁带宽度是固定的,种类数量不多.后者则需要引入杂质,杂质能级在禁带内被产生,主要由引入的杂质能级导致发光,占闪烁晶体的主要部分.[12]这两类发光机理如图1 所示,区别就在于杂质所引入的陷阱能级是否参与电子或空穴的捕获.
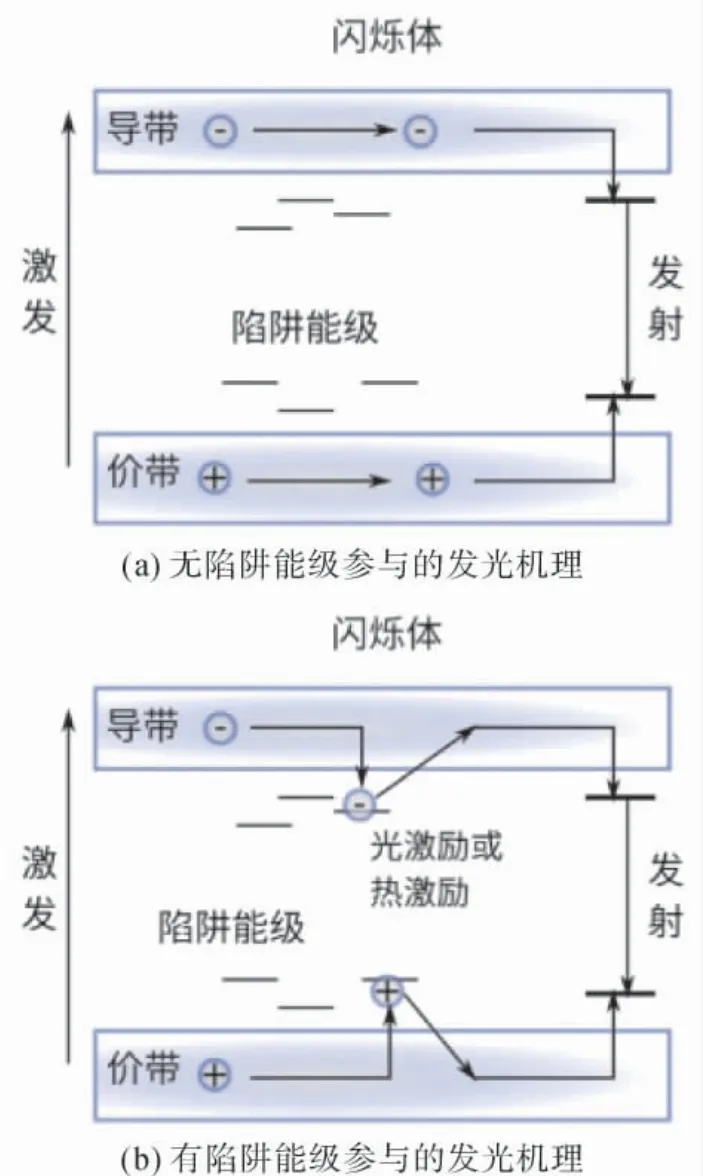
图1 闪烁晶体发光原理示意图Fig.1 Schematic diagram of scintillation crystal
不难发现,缺陷能级的捕获会延迟发光的时间,形成余辉.但在实验研究中,由于包括氧空位在内的多种缺陷是共存的,在解释余辉现象时,无法区分陷阱能级的来源,也无法有针对性地改进.从计算的角度切入可以对单一缺陷进行研究,更有针对性.
由于f 层电子的强关联效应,目前计算材料学常用的密度泛函理论需要引入修正才能近似描述稀土氧化物的能带结构,因此关于RE2O3: Eu( RE=Lu,Y,Sc) 型闪烁晶体的计算建模类型的文献起步较晚.近年来,关于RE2O3( RE=Sc,Y,Lu) 型倍半氧化物的计算研究初见成效.2009年,Cui zhifeng 等人通过PBE +U 计算了Lu2O3和Y2O3的能带宽度[13].2016 年,Sonu Sharma 等人使用修正的含局域密度近似( Local Density Approximation: LDA) 方法对Sc2O3和Y2O3进行第一性原理计算[14],得出了禁带宽度的值.本文将使用第一性原理计算,通过PBE +U 的方式对含氧空位的RE2O3( RE=Sc,Y,Lu) 型倍半氧化物进行计算,并从微观角度对缺陷能级进行解释,为实验提供理论依据.
2 计 算
密度泛函理论会计算出所有宿主原子的弛豫状态,将原子位置函数的总能量取最小化值后的得到一个稳定的结构.大多数泛函在密度泛函理论体系下的LDA 计算均可得到合理误差范围内的结果[15].
对于强关联体系,可用LDA +U 的方法进行修正( 在半芯态增加一个额外的库伦相互作用力U) ,在LDA + U 中的电子被分离成局域化的电子,这样通过Hamiltonian 模型中的哈伯德项把库仑斥力U 考虑在内,离域或者巡游电子在LDA 中可通过常用的轨道依赖单电子电势能被很好的描述[15].LDA+U 方法最初用来解决局域化轨道中的电子,重点是开壳层体系( 具有强关联的体系) ,如过渡金属化合物和稀土金属及化合物.
使用LDA+U 的U 值的实际含义是额外添加了一个能量U,包含U 势的LDA +U 会导致太多的全空/全满占据态,虽然可以从一定程度改善禁带宽度,但是会存在一定误差.
对于RE 元素,我们选择4f 电子作为核心电子生成的PAW 数据集.对于Eu,我们选择了由完全4f 价电子生成的PAW 数据集.氧的PAW 数据集是使用1.52 玻尔的核心半径和部分核心修正生成的.Lu、Y、Sc、Eu 和O 的所有PAW 数据集都来自VASP[16]代码附带的PAW 数据集库.平面波基的截止能量为500 eV,采用高斯涂抹法模拟电子态占据,涂抹宽度为0.05 eV.第一个布里渊区在根据Monkhorst -Pack 方法[17]生成的特殊k 点上采样,使用所有系统( 包括纯和掺Eu 的RE2O3) 的网格,自洽场迭代的标准设置为1.0 ×10-6eV.在结构优化中,每个原子上的残余力减小到0.005 eV 或更小.
3 计算结果
RE2O3( RE=Lu,Y,Sc) 作为基体材料的闪烁晶体虽然近年来已有多项实验研究,现已知余辉性质与能带结构密切相关,尤其是杂质能级在半导体的禁带中的位置.因此晶体结构对能带结构的影响具有决定性因素,建立合适的模型就至关重要.
3.1 RE2O3晶体结构
我们从aflow.org[18]的晶体结构数据库中下载了初始模型,考虑到结构的合理性,使用vasp 对结构进行弛豫优化计算,优化后的结构会考虑到离子相互作用、点缺陷造成的相邻位置塌陷等因素,更倾向于实际值.优化后的晶格常数如表1所示:
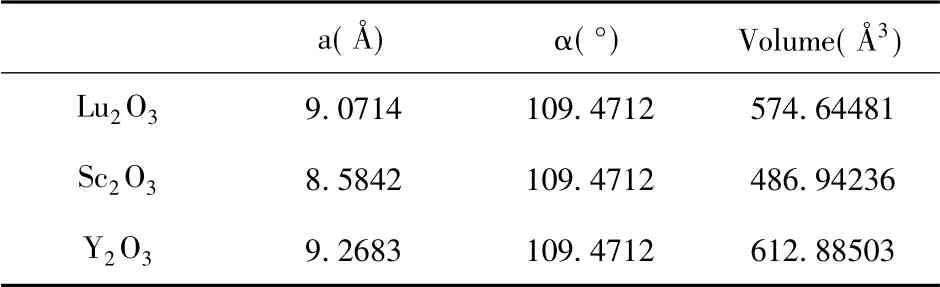
表1 优化后的RE2O3晶格常数( 立方晶系,a=b=c,α=β=γ)Table 1 Optimized lattice constants of RE2O3( cubic system,a=b=c,α=β=γ)
在计算中,我们使用 Brich - Murnaghan( BM) 状态方程对不同体积下的EOS( Equation of States) 进行分析.EOS 包含V0( 最低能量时的平衡体积值) ,E0( 平衡能量值) ,B0( V0的体弹性模量) ,B’( B0的一阶导数) 四个参数,相对应的公式为如公式1 所示,拟合BM 状态方程所用的参数如表2 所示.
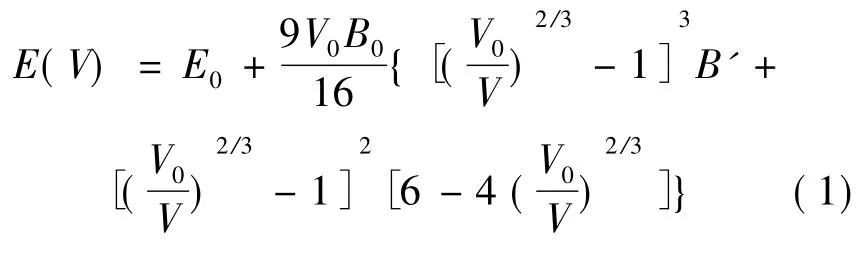
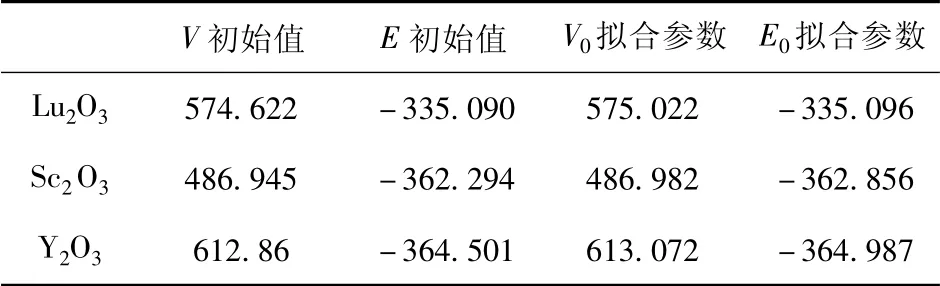
表2 BM 状态方程拟合参数及初始参数( 能量单位为eV,体积单位为Å3)Table 2 Fitting parameters and initial parameters of BM equation of state( E in eV and V in Å3)
图3 展示了RE2O3( RE=Lu,Y,Sc) 的体积-能量变化关系图,体积由0.85 倍到1.03 倍变化.不难发现,在三种材料随着体积的变化,分别在接近VLu=575.02 Å3,VSc=486.98 Å3,VY=613.07 Å3处拟合曲线达到最低值.表明体系的结构稳定值为上述值,建模可靠.
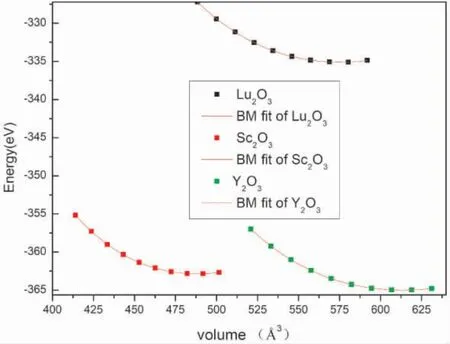
图2 不同体系的RE2O3的状态方程Fig.2 Equation of state of RE2O3 in different systems
3.2 RE2O3含氧空位的晶体结构
在实验制备的过程中,由于各种不可控因素,或多或少都会引入一些缺陷.而存在较多的,就是由气氛环境所导致的氧空位缺陷.虽然缺陷的比例可能会很小,但也足以对发光性能造成一定的影响.为了核实氧空位带来的影响,我们将缺陷放大进行计算.
在80 个原子的立方晶胞中( 含32 个RE 原子,48 个氧原子) ,若挖去1 个氧原子,占比1/48,约2%.对于氧空位而言,数值已经不小了.起初,我们纠结于究竟挖去哪个氧原子,毕竟氧原子的缺失会影响结构的塌陷.经过对堆垛结构( 图3) 的反复研究后发现,不论挖去哪个氧原子,由于对称性和周期性,其实影响S6 和C2 的数量是一样的.我们挖去了如图4 位置的氧原子,结构模型和初始晶胞对比情况如图所示:

图3 沿( 001) 晶面的各层原子的排列情况( 以RE2O3为例)Fig.3 The arrangement of atoms in each layer along (001) crystal plane
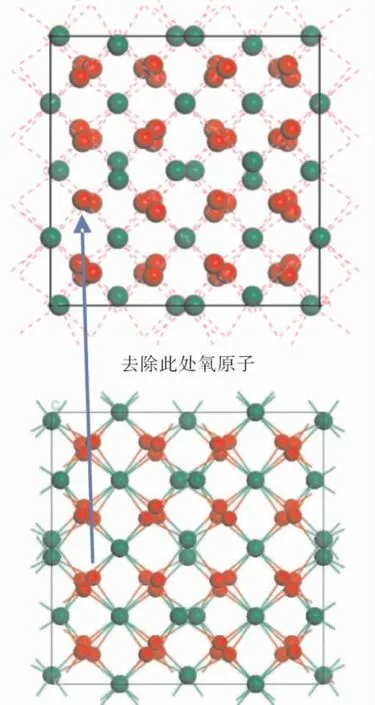
图4 存在氧空位( 上) 和原始晶胞( 下) 结构建模图Fig.4 Oxygen vacancy ( up) and primitive cell( down) structure modeling
同样,我们分别对含氧空位的Lu,Y,Sc 三种体系进行了结构弛豫优化.为了直观地反应其氧空位形成的难易程度,我们根据公式( 2) 列出了表3:
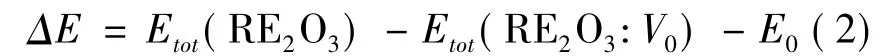

表3 三种体系氧空位的形成能对比( 单位: eV,使用pbeu 计算,单个氧原子形成能为-4.925 eV)Table 3 Comparison of formation energies of oxygen vacancies in three systems( unit: eV)
从表3 可见,Y2O3中氧空位的形成能最高,氧空位最难形成; Lu2O3中的氧空位形成能最低.三者数值相差不多,发生氧空位的概率相近.
3.3 PBEU 计算的能带结构
在计算中,我们提到PBE 被广泛应用,但是其结果在应用于能带结构的时候,存在误差.PBE+U 可优化计算结果,可以达到一个更加精确的值.
图5 展示了用PBEU 计算的RE2O3能带结构,可以发现三种材料非常类似.禁带宽度都为4eV左右.对比实验值( Lu2O3gap=5.5 eV[19],Sc2O3gap=5.7 eV[20],Y2O3gap=4.54 eV[21]) 确实存在一定误差.其他文献中计算值为Sc2O3gap=3.84 eV,Y2O3gap=4.05 eV[14].计算上的误差是由于f 轨道上的电子既有局域化的( localized) ,也有游离的( Itinerant) ,所以单纯的+U 修正不能描述f轨道的全部电子.
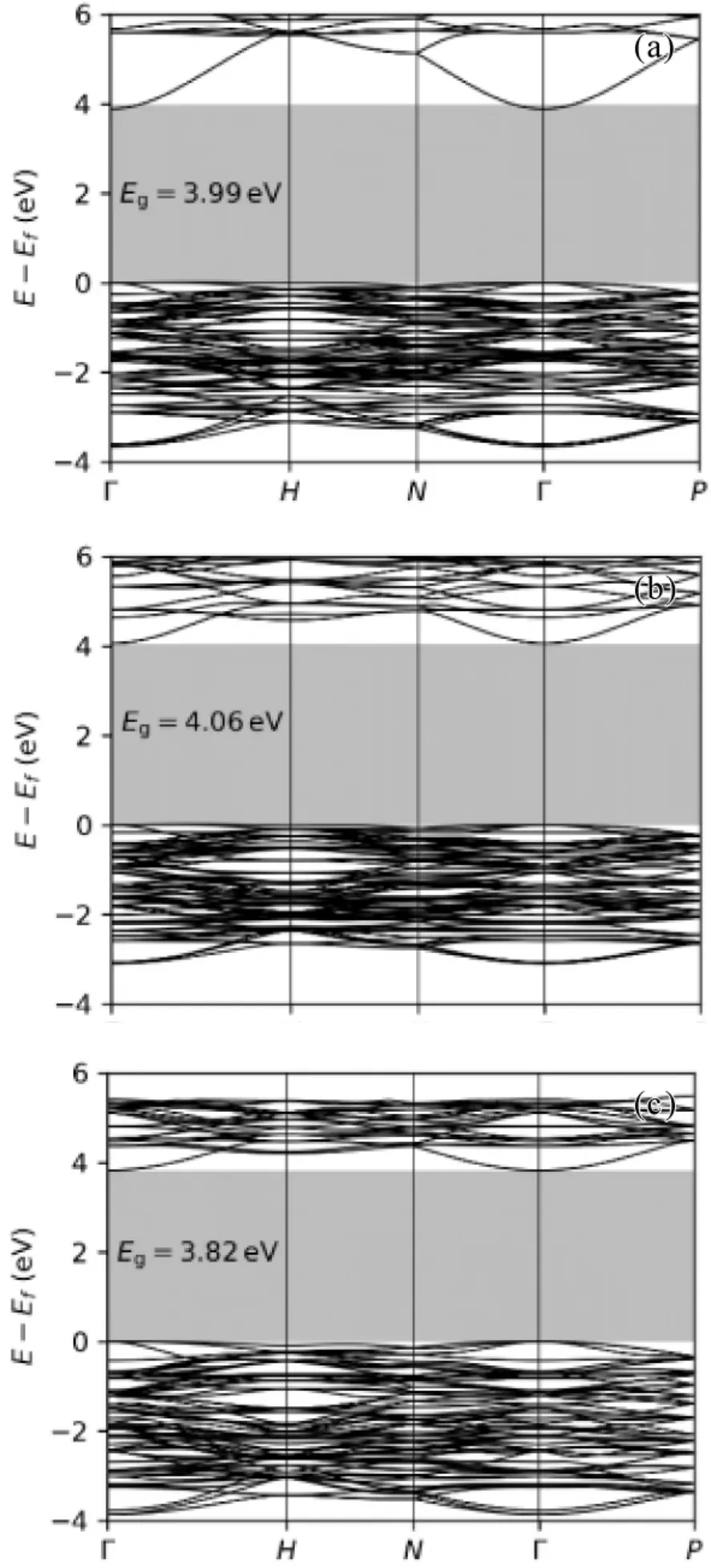
图5 用PBEU 分别计算了( a) Lu2O3( b) Y2O3 和( c)Sc2O3的价带和低能导带结构.阴影区域表示价带最大值( VBM) 和导带最小值之间的带隙.子图中还显示了带隙的值.( a) 用PBEU计算的Lu2O3 能带结构( b) 用PBEU 计算的Lu2O3能带结构( c) 用PBEU 计算的Sc2O3 能带结构Fig.5 The valence band and low energy conduction band structures of ( a) Lu2O3( b) Y2O3 和( c)Sc2O3 are calculated by PBEU.The shaded area represents the band gap between the maximum valence band ( VBM) and the minimum conduction band.The value of the band gap is also shown in the subgraph.
3.4 存在氧空位情况下PBEU 计算的能带结构
在RE2O3( RE=Lu,Y,Sc) 型闪烁晶体中的制备过程中,氧空位的形成是很难避免的.氧空位的分布也决定着C2 和S6 格位的分布变化,进而影响这些材料的能带结构.特别是当化学配比发生变化时,氧空位的增减也会导致氧空位分布的变化.因此我们将建立带有氧空位的结构模型,使用PBE+U 进行计算,查看其能带结构带来的趋势变化.
在Sc2O3: VO体系中( 图6a) ,我们可以发现,在禁带中间出现了一条新增的能带.这条能带很好地阐述了氧空位带来的影响: 由于氧离子带负电,氧空位带正电,因此会捕获电子.在图上禁带中约2.3 eV 处存在较多( 能带线越平整代表能态密度越大) 的电子陷阱.陷阱的增多,会导致捕获的增多,也就意味着余辉将会增强.根据能量守恒可知,发光强度将会减弱.因此,在Sc2O3: VO体系中,我们并不希望引入氧空位.

图6 RE2O3: 2%VO的能带图Fig.6 Band structures of RE2O3: 2%VO
在Y2O3: VO体系中( 图6b) ,除了和Sc2O3:VO体系的相似之处外.我们还发现在导带底下方不远处新增了一条能带.该能带会导致更多的电子被捕获,从而增强余辉效应.氧空位对于Y2O3: VO体系的影响会大于Sc2O3: VO体系.
Lu2O3: VO体系( 图6c) 和Y2O3: VO体系非常相似,但在约2.1 eV 处的能带线较为曲折,说明能态密度并没有Y2O3: VO体系中的这么大,电子陷阱没有这么多.
虽然在三种体系中,氧空位都会引入陷阱态,增强余辉效应.但是影响的强度不同,依次为:Sc2O3<Lu2O3<Y2O3.也就是说,制备工艺对于Y2O3的影响是最大的,对于Sc2O3的影响是最小的.这也就很好地解释了,在实验中Lu2O3发光强度会优于Y2O3这个现象.而Sc2O3由于结构的细微差异(4.2 节) ,在低掺杂情况下,发光强度不如Lu2O3和Y2O3.
4 结 论
通过计算建模和对BM 状态方程的分析,我们确定了这三个体系的结构模型.计算得出其能带结构非常相近.
在引入氧空位的情况下,通过计算形成能发现氧空位发生的难易程度相近.在Y2O3中的氧空位会引入最多的缺陷能级,导致余辉效应增加,发光效率降低,其最容易受到氧空位影响,Lu2O3其次,Sc2O3受到的影响最低.希望能为接下来的实验研究提供理论依据.
猜你喜欢
南昌工程学院学报(2022年4期)2022-12-18
椰城(2021年12期)2021-12-10
北京航空航天大学学报(2021年7期)2021-08-13
少儿科学周刊·少年版(2021年22期)2021-01-17
科技创新与应用(2020年8期)2020-03-13
浙江工业大学学报(2019年6期)2019-11-01
陶瓷学报(2019年5期)2019-01-12
科技创新与品牌(2018年9期)2018-10-24
腐蚀与防护(2016年7期)2016-09-14
导航定位学报(2015年2期)2015-06-05
