
MRE11 基因变异致共济失调毛细血管扩张样障碍1 型1 例报告
2022-05-16 04:54贾倩芳崔清洋周福军
临床儿科杂志 2022年5期
贾倩芳 崔清洋 周福军
新乡医学院第一附属医院儿科(河南卫辉 453100)
共济失调毛细血管扩张症(ataxia-telangiectasia,AT)为一类罕见累及神经、血管、皮肤及内分泌等多系统的常染色体隐性遗传性疾病,发病率为1/100000~1/40000,ATM基因变异为其唯一的致病病因,该病典型临床表型为婴幼儿期发病的进行性小脑共济失调,眼球结膜和皮肤毛细血管扩张,免疫缺陷,反复发作的副鼻窦炎及肺部感染[1]。共济失调毛细血管扩张样障碍(ataxia telangiectasia-like disorders,ATLD,OMIM #604391),是由Stewart在1999年首次提到[2],临床特征与AT患者非常相似,为AT 一种罕见变异,ATLD 患者表现为进行性小脑共济失调、眼球运动不能和认知缓慢恶化,且神经学特征起病晚,进展慢,也无发病率报道,目前全球共报道ATLD 患者40 余例。而ATLD 与AT 相比,无毛细血管扩张、甲胎蛋白升高和免疫球蛋白水平降低等神经系统外特征[3]。
AT 和ATLD 均属于染色体不稳定综合征,其特征是染色体不稳定和对放射性敏感性增加,染色体不稳定综合征是一组与染色体不稳定和断裂有关的遗传性疾病,为自发或对DNA 损伤剂反应所致[4]。ATLD 与两种不寻常遗传病相关:ATLD 1 和基因MRE 11有关,而ATLD 2 与基因PCNA变异有关[5]。本文回顾性分析1例ATLD1患儿的临床资料及基因检测结果,以期加强儿科医师对ATLD1疾病的认识。
1 临床资料
女性患儿,11岁,汉族,无家族遗传史,因间歇左膝关节疼痛3 年余,运动、智力倒退2 年余入院。患儿系领养儿,G2P2,足月顺产,无产伤及窒息史。正常饮食,生活方式无特殊,现为小学四年级,学习成绩差(主要表现为语文阅读理解及数学理解能力低下),话语缓慢。生物学父母均体健,非近亲结婚。本次入院前门诊予以热疗后左膝关节疼痛减轻,智力倒退加重不明显,运动倒退明显,但左膝关节疼痛易反复。本病例的发表及相关研究均获得监护人知情同意。患儿诊治时间轴见表1。

表1 患儿诊治时间轴
体格检查:头围50.5 cm,身高127 cm,眼球运动不能,四肢肌张力可,四肢肌力IV级,膝腱反射未引出,双手轮替试验阳性,闭目难立征阳性,指鼻试验阳性,跟-膝-胫试验弱阳性,左侧踝阵挛弱阳性。专科检查:核心力量差,协调性差,骨盆控制性差。步行:膝关节与踝关节不稳,身体重心左右摇摆,呈轻度“X型腿”,“外八字”步态。支撑相前期:该患儿双侧踝关节背屈不充分,在承重反应期和支撑相中期时,双侧膝过伸,并伴有“外八字”步态,左侧足外翻,右足前脚掌外侧负重。摆动相中期:膝关节僵硬,双侧髋关节轻微内旋,膝关节伸展不充分,并伴有膝外翻。骨盆左倾,双膝外翻,双膝过伸。
面对五彩斑斓的西域山川,有许多画家进行了色彩的大胆尝试,这也不愧为一种艺术表现形式的探索。尤其是冰川与绿树黄花同时映入我们的眼帘时,作为“视觉动物”的艺术家真的很难经得住这种色彩美的巨大诱惑。但人们在表现山川之“象”时,却不能仅仅停留在“像”的层面上,是中国画,就需要考虑格调的问题。冰山与烈焰如何兼具一身?是以色貌色还是以形写神?视觉的巨大冲击需要以思想的高度升华为转机,才能演变为山水之境的历史重生。
MRE 11基因定位于11 q 21,基因组全长约74 kb,包含19个外显子,外显子长度约2 588 bp,编码680个氨基酸的Mre11蛋白。目前业已报道MRE11基因变异约为779种(https://www.ncbi.nlm.nih.gov/clinvar/),包括移码变异34种,错义变异604种,无义变异22种,剪接位点变异35种,非翻译区变异84种,以错义变异最常见。最常见报道为复合杂合或纯合错义变异,其中一些变异可能不会改变翻译的氨基酸,但会影响蛋白质效率。本例患儿为错义变异组成的复合杂合变异,支持文献报道。

图1 患儿头颅MRI 表现
测序分析发现:患儿MRE11基因第8外显子c.728 G>C(p.W243S)和第3外显子c.68 T>C(p.L23P)错义变异,上述变异可能造成蛋白质功能遭受严重影响,上述两变异位点均未见文献报道(所参考数据库 HGMD Pro及 PubMed)。c.728 G>C和c.68 T>C两变异不属于多态性变化,在人群中发生频率极为低下(所参考的数据库 1000Genomes和dbSNP)。因患儿系领养儿,生物学父母拒绝采血,未能家系验证变异来源,见图2及图3。
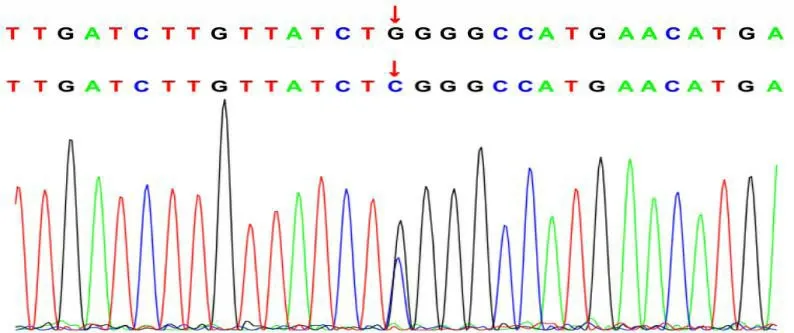
图2 MRE11 基因c.728 G>C 测序峰图
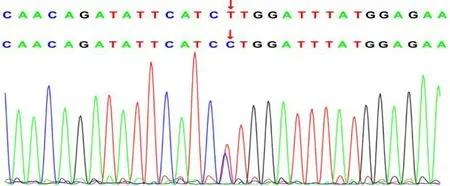
图3 MRE11 基因c.68 T>C 测序峰图
结合患儿临床表现、实验室检查、头颅MRI 及基因检测分析诊断MRE 11基因c.728 G>C 及c.68 T>C复合杂合变异所致的ATLD1基本明确,告知患儿家属该病目前尚无特效治疗,建议患儿定期返院康复治疗,于7个月后因左膝关节疼痛,运动及智力倒退再次入院,予电子生物反馈(胫前肌 股四头肌)、低频脉冲治疗、关节松动训练、牵引、作业疗法、手功能训练、运动课等康复训练,治疗后左膝疼痛减轻,但智力倒退无明显加重。出院后随访至今,疼痛仍易反复,智力倒退加重不明显,运动倒退明显,未再康复治疗。
根据美国医学遗传学与基因组学学会(ACMG)联合美国分子病理学会(AMP)2015年制订的基因序列变异标准和指南进行致病性分析[6]。MRE 11基因c.728 G>C 位点的证据强度为“PM 1+PM 2+PP 3”,判断为导致受检者发病的临床意义不明的变异。MRE 11基因c.68 T>C 位点的证据强度为“PM 2+PP 3”,判断为临床意义不明的变异。虽MRE11基因c.728 G>C及c.68 T>C错义变异均判断为临床意义不明变异,尽管目前尚无文献报道且判断为临床意义不明,但亦未有无致病性的定论,需待日后进一步行相关验证。
2 讨论
韦氏智力测试量表:言语测试IQ 50,操作测试IQ 54,检验结果IQ 47分,提示中度智力低下。实验室检查:血常规、肝肾功能、心肌酶、血氨、RF、ASO及甲状腺功能均未见异常,电解质:钙2.0 mmol/L(正常范围2.08~2.80 mmol/L),稍偏低,余无异常;乳酸2.3 mmol/L(正常范围0.5~1.5 mmol/L),稍偏高;免疫球蛋白A、免疫球蛋白G及免疫球蛋白M和淋巴细胞亚群绝对计数均未见异常;抗ENA抗体谱未见异常;甲胎蛋白未见异常;尿气相色谱-质谱提示氨基酸尿伴酮症和糖尿;头颅MRI示双侧小脑半球脑沟加深,提示小脑萎缩(图1)。患儿入院后暂未予治疗,因患儿智力、运动发育倒退,且神经系统检查提示共济失调,不能排除遗传性疾病(线粒体脑肌病?异染性脑白质营养不良?)可能。经医学伦理审核及家属知情同意后,采集患儿和父母外周血送检基因检测。
在建立合适的RAPD反应体系的基础上,从50个10 bp随机引物中选用最好的引物S11、S13、S29、S32、S40、S43、S44、S47、S49、S50 用于正式扩增,对 6个供试的三叶木通叶片基因组DNA扩增出10张指纹图谱,共获得86条DNA谱带,其中73条多态性带,占总扩增带数的84.9%,每个引物扩增的带数在7~11条,平均为8.6条,扩增出的DNA片段大小在100~1 000 bp,表3为所选的10种引物扩增结果。
经文献检索,目前全球共报道42例ATLD患者,其中38例为ATLD1,4例为ATLD2。ATLD1临床特点是小脑共济失调、眼球运动异常、放射敏感性、舞蹈性手足徐动症和肌张力障碍。在ATLD1中很少发现身材矮小、小头畸形、认知障碍、面部运动障碍和神经病变。ATLD1最常见的动眼神经异常是缓慢和不规则眼跳、动眼神经失用症、会聚延迟和注视诱发的眼球震颤。ATLD2最早于2014年发现,虽1986年之后发现其在DNA 修复和PCNA 基因复制中的功能。ATLD2临床特征包括小脑共济失调、身材矮小、听力丧失、早老、毛细血管扩张和光敏性。其他临床症状包括智力障碍、足部畸形、话语和吞咽困难及肌无力。动眼神经异常偶有报道。与AT相反,ATLD发生恶性肿瘤风险可能较低;但ATLD1中报道肺腺癌,ATLD2中报道阳光诱导皮肤癌。Taylor 等[8]总结AT、ATLD 1 和ATLD 2 临床特征差异。该患儿有小脑共济失调、眼球运动异常、身材矮小、小头畸形及认知障碍和左侧踝阵挛,但无肌张力障碍,无毛细血管扩张、甲胎蛋白升高和免疫球蛋白水平降低等神经系统外特征,支持文献报道。
ATLD 1 和AT 早期头颅MRI 可能正常。但多数ATLD1患者病程中表现为小脑萎缩,主要在小脑蚓部,我们报道本例患儿小脑萎缩在小脑蚓部,支持文献报道。
“雅虎”搜索引擎的核心是Anthelion 爬虫技术,使用Nutch 算法实现。Anthelion 爬虫的评分机制可同时为每个链接网页评分,将结果提供给分类器进行分析,分类器可调整优化,理论上能够获取较高的查准率;其信息解析机制可针对网页内容提取语义数据,从网页中获取数据、格式和注释信息,并存储于内容字段中,将其置为特征信息量;可存储新字段加入索引以扩大查询范围。实验中使用雅虎搜索引擎在互联网中搜索结果并不理想,其主要原因在于爬虫中分类器的优化策略与专业关键词的匹配度存在差距,其整体设计目标适应的范围更广。
虽尚无基因型与临床表型关系报道,但Fernet等[7]报道4例英国和2例意大利ATLD1患者中,2例携带MRE11基因纯合变异,导致C端截短的Mre11蛋白表达,缺失最后76 个氨基酸。另外4 例患者为复合杂合变异,表达全长变异Mre11蛋白,表现为较温和临床表型,故纯合变异较复合杂合变异临床表型症状重,该患儿为MRE11基因c.728 G>C及c.68 T>C 复合杂合变异,临床表型症状较轻,支持文献报道。
目前ATLD1治疗无特异性,均为对症治疗。Ser等[9]报道2例ATLD1成人患者,其中33岁女性先证者因双侧上下肢及口腔肌张力障碍,给予左旋多巴(900 mg/d)后步态障碍得以改善,表现为无帮助下步行距离增加及流涎减少,左旋多巴1 年余治疗中未发现运动并发症,但剂量渐减少至300 mg/d。
尽管尚无法明确MRE11基因c.728 G>C及c.68 T>C变异位点致病性,但患儿有ATLD1典型临床表现,且c.728 G>C及c.68 T>C变异(尽管目前c.728 G>C 尚无文献报道且c.728 G>C 及c.68 T>C 均判断为临床意义不明,但亦未有无致病性的定论,需待日后进一步行相关验证。综合考虑基因结果及患儿临床表型,基本确认MRE11基因c.728 G>C及c.68 T>C变异位点为患儿致病性变异。
本文以宁波市主城区为研究对象,利用SPSS软件对重分类后的POI数据的街区密度进行主成分分析,然后根据各主成分对原始指标的载荷情况选出能够表示商业区、文教区、工业区的综合指标,即以第一主成分作为反映商业区特征的综合指标,以第二主成分作为反映文教区特征的综合指标,以第五主成分作为反映工业区特征的综合指标,对这三个综合指标进行可视化表达(图一至图三),颜色越深代表该综合指标载荷的相应POI点的数量越高,就越能够识别相应的功能区。依据可视化表达结果分析宁波市主城区功能区分布格局:
导致儿童运动发育落后和智力障碍的病因较多,及时基因检测可及早明确病因、产前咨询及母亲再孕时产前诊断,避免过度治疗及出生缺陷。本例患儿为国内首次报道,且MRE11基因c.728 G>C及c.68 T>C变异位点国际上尚未报道,扩充了ATLD1国际基因变异谱。
猜你喜欢
分子催化(2022年1期)2022-11-02
分析测试学报(2022年9期)2022-09-21
中国农业科学(2022年16期)2022-09-19
中国典型病例大全(2022年12期)2022-05-13
支部建设(2020年15期)2020-07-08
好孩子画报(2019年8期)2019-09-19
电脑知识与技术(2018年19期)2018-11-01
哲思2.0(2017年12期)2017-03-13
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
