
光、电催化CO2耦合有机化合物催化转化研究进展
2022-09-13 01:03王若愚李金昊宁晨君赵宇飞
洁净煤技术 2022年9期
王若愚,李金昊,宁晨君,田 强,赵宇飞
(1.北京化工大学 化工资源有效利用国家重点实验室,北京 100029;2.北京化工大学 化学学院,北京 100029;3.山东万新威纳材料科技有限公司,山东 临沂 276000)
0 引 言
现阶段,化石能源的过度使用导致温室气体CO2大量排放,造成了一系列生态环境问题。基于目前“碳达峰、碳中和”目标,开展CO2资源化路线,发展高效的催化转化技术,是碳中和技术研发的重要内容,也是建立清洁安全高效能源体系的重要途径之一,对于实现双碳目标具有重要意义。

笔者综述了近几年来光、电催化CO2与有机化合物耦合方面的研究进展,围绕其反应机理,即有机物的活化和自由基的生成展开论述,讨论反应效率及其在合成与生产中的应用,并总结了各类催化的特点和优势,指出反应中可能存在的问题并对该领域未来发展方向进行了展望。
1 太阳光驱动的CO2耦合
太阳能光驱动反应主要分为光催化反应、光热反应和光电反应等,本节重点讲述应用于CO2耦合的光催化和光热催化。光催化反应中,由于太阳能的激发,催化剂表面产生电子和空穴。扩散到催化剂表面的载流子能量很高,进一步注入反应分子中,可产生高反应活性的物种。而光热反应通过光能到热能的转化,提高了反应体系温度,从而驱动反应进行。因此,光驱动可使热力学不利的反应在温和条件下进行。通过对光能的直接利用,可以缓和、甚至避免热催化CO2反应在固碳的同时产生大量碳排放问题,为光驱动CO2耦合提供新思路。
1.1 CO2与醇的光催化脱水缩合


(1)
(2)
(3)
(4)
这项工作并未将反应条件降至足够温和,但证明通过光催化可能使该反应成为理想的绿色化学反应,为此类反应催化剂的设计奠定基础。目前,光催化CO2和醇类的缩合反应还存在产率偏低、反应速率较慢等问题,相关研究还需进一步开展。
1.2 CO2与环氧化物的光(热)催化转化

图1 CO2环加成反应机理Fig.1 Mechanism of CO2 cycloaddition reaction
由于该反应是吸热反应,通常在40 ℃以上,甚至加压条件下才能进行[15-17]。目前大多数热催化体系都通过调控催化剂表面的Lewis酸碱来降低环氧开环的活化能,具有光热效应的催化剂可以在光照而非直接加热的条件下为体系提供热量,因而对该反应催化活性较好,催化性能优越。近几年该反应中一些光催化剂性能及光照条件见表1。
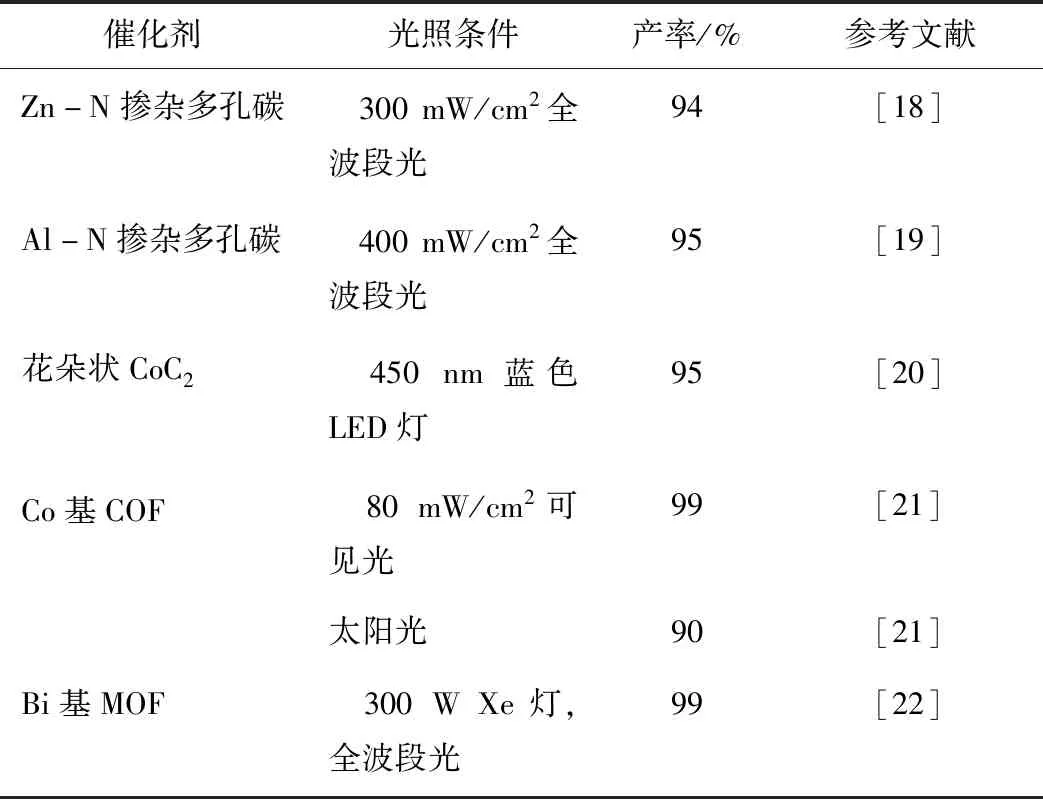
表1 CO2与环氧化物加成中一些光催化反应条件和效率Table 1 Conditions and efficiency of some photocatalytic reactions in the cycloaddition of CO2 with epoxides

图2 一些CO2环加成反应的光催化剂[18,20]Fig.2 Some photocatalysts for CO2 cycloaddition reactions[18,20]
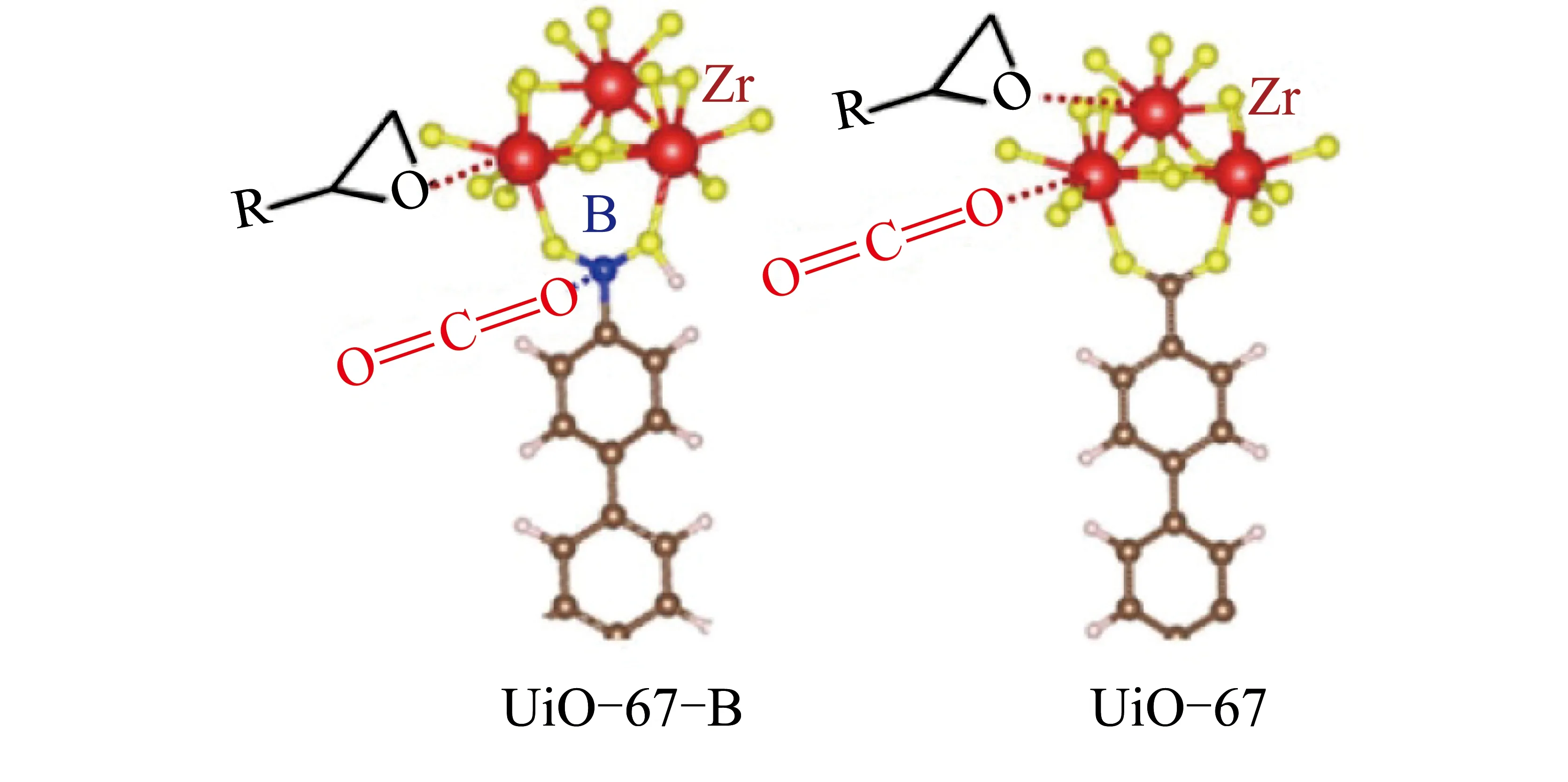
图3 环氧化物和CO2在UiO-67和UiO-67-B上的吸附[25]Fig.3 Adsorption of epoxide and CO2 over UiO-67 and UiO-67-B[25]
目前,太阳能驱动的CO2对环氧化物的环加成反应以多相催化为主,具有产率高、可循环高效利用、反应条件温和、原子经济性高等优点,且相关反应机理研究较深入。但存在光能利用效率较低,需在大功率全波段光照下才能达到较高反应速率等问题,因此如何提高该反应对光的利用率仍需进一步研究。
1.3 C—H键的光催化羧化
将CO2插入C—H键生成对应的羧酸,可用于有机合成和CO2转化。传统羧化方式分为2种:① 配合物催化,即在氧化剂存在下,由过渡金属配合物催化CO进行反应;② 直接利用CO2进行反应,但是需在高温高压和强碱等较剧烈的反应条件下进行,或利用活泼金属镁、锂等将底物制成高度活泼的金属有机试剂,再与CO2发生羧化反应。这些反应危险性较高,或反应条件过于剧烈,能源消耗较高。近年来,温和条件下C—H键与CO2的羧化引起关注,然而,C—H键键能较高,均裂、异裂难以活化。热催化下该反应大多需要100 ℃以上才能顺利进行[26],光热效应催化剂难以达到该温度,因此寻找其他合适的催化策略使C—H键羧化高效进行是研究重点。系统论述了均相分子型催化剂和多相半导体型催化剂对C—H键的光催化羧化反应,以及一类具有特殊结构的有机物分子的光化学羧化反应。光催化C—H键羧化的反应条件和催化性能见表2。
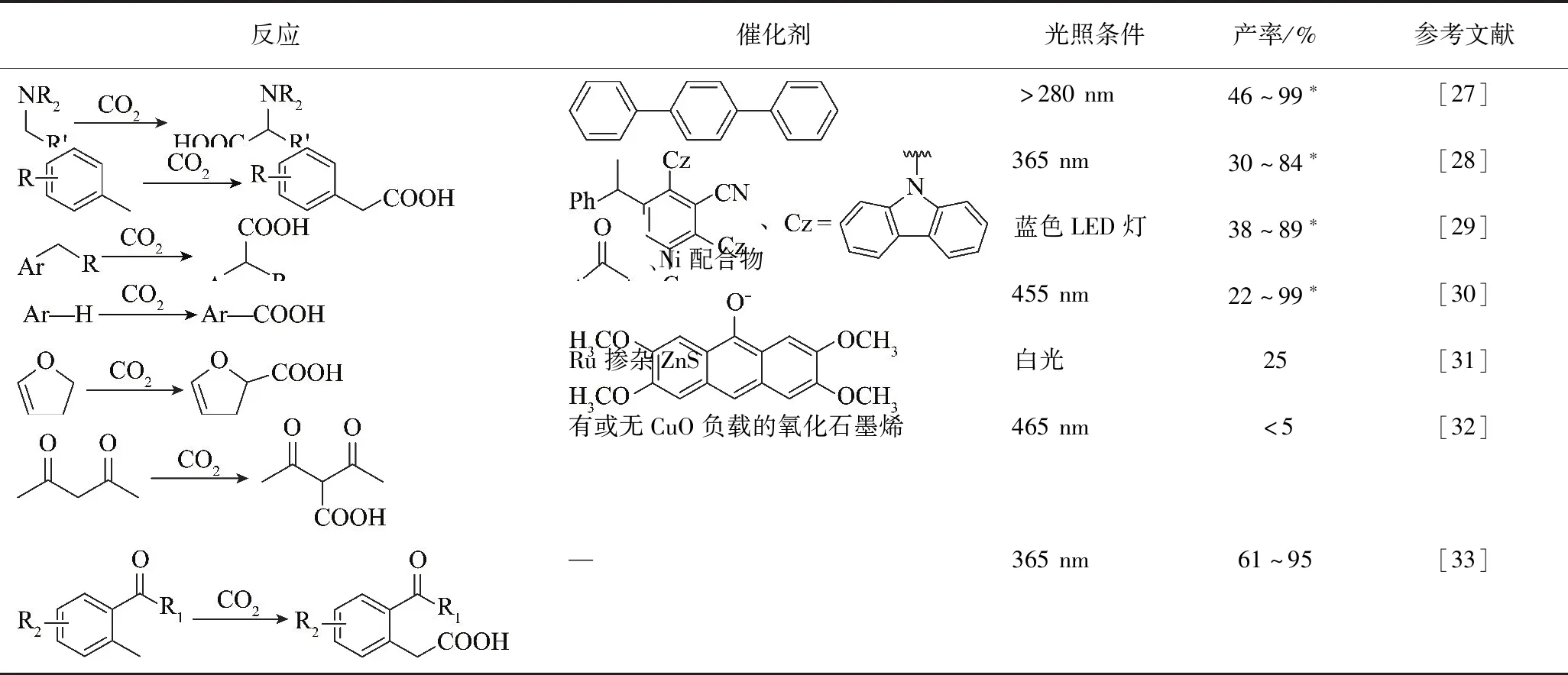
表2 CO2与C—H键羧化中一些光催化反应的条件和效率Table 2 Conditions and efficiency of some photocatalytic reactions in the carboxylation of CO2 and C—H bond
1.3.1 均相催化的C—H键羧化
均相小分子催化剂在光照下激发,可以生成高度活泼的激发态分子,通过其与反应物分子之间电子或原子的转移,实现C(sp3)—H键羧化。由于π—π*跃迁具有较高的吸光系数,且通常位于近紫外和可见光区,因此现阶段研究主要选择具有大共轭体系的有机分子进行相关反应。研究人员对均相催化C—H键羧化的反应机理进行深度探究,发现受催化剂自身性质影响,激发态的催化剂分子可能夺取氢原子或电子,进而获得具有高度反应活性的自由基,进攻CO2分子实现有机物的羧化反应。不同催化剂作用下自由基生成方式如图4(a)~4(c)所示(SET为单电子转移,HAT为氢原子转移,cat.为催化剂)。芳烃作为催化剂时,其激发态分子为无明显定域分布的孤电子,以夺取电子为主。若底物中有n电子,激发态分子将夺取底物中的一个n电子,此时邻位质子的酸性大幅增强,被碱夺取后生成自由基[27];若底物中没有n电子,需加入有n电子的助催化剂(如硫醇)协助电子转移,夺取底物中的氢原子,使底物成为自由基[28]。酮作为催化剂时,其激发态分子中存在定域分布在氧原子处的孤电子,此时激发态的催化剂分子可直接夺取底物中的氢原子,生成自由基中间体[29]。上述自由基的产生是C(sp3)—H键羧化反应的关键步骤,通过其对CO2分子的进攻,导致最终产物羧酸(盐)生成(图4(d))。
与C(sp3)—H键相比,C(sp2)—H键键能更高,导致激发态分子对C(sp2)—H键的羧化更难。基于此,SCHMALZBAUER等[30]发现C(sp2)—H键的光催化羧化反应机理与C(sp3)—H键羧化相反(图4(e)),主要通过先加成、后消除方式将CO2插入C—H键,即反应物由激发态的催化剂分子获得电子,生成具有较强亲核性的自由基阴离子,并进攻CO2导致(共轭)π键的短暂破坏,进一步通过碱对质子的去除,使(共轭)π键恢复并生成产物羧酸(盐)。

图4 C—H键均相光催化羧化的部分机理[27-30]Fig.4 Some mechanism of homogeneous photocatalytic carboxylation of C—H bond[27-30]
均相催化的C—H键羧化反应与相应的热催化反应相比,催化路径更加温和、安全和高效。用于合成时,该反应具有明确的反应机理,可广泛用于苄基氢、烯丙基氢等活泼氢及芳基氢的羧基化反应。但对CO2固定而言,该方法产率和选择性受底物影响较大,且通常需使用能量较高的蓝紫光及近紫外光,对太阳光利用率低,在应用中受限。
1.3.2 多相催化的C—H键羧化

图5 乙酰丙酮光催化羧化的可能机理[37]Fig.5 A possible mechanism for the photocatalytic carboxylation of acac[37]
1.3.3 无需催化剂的光化学C—H键羧化反应
一些特殊的有机分子可以在无催化剂的条件下直接与CO2发生光化学反应,进而获得相应的羧酸。MASUDA等[39]报道了一类邻烷基苯基酮与CO2的光催化羧化反应(图6),在常温常压下,对合适的底物收率可达95%。受光照产生的激发态邻烷基苯基酮分子发生分子内氢原子转移,退激后生成Z型或E型烯醇式异构体,前者互变异构返回酮式结构,后者与CO2发生[4+2]环加成反应,开环后生成产物羧酸。该反应条件温和,无需催化剂且反应高效,虽然对底物结构的特殊要求使其难以普遍应用,但在该反应中,CO2分子可视为缺电子亲双烯体,烯醇式中间体可视为富电子双烯体,这意味着CO2分子的π键可直接参与周环反应,为CO2应用开辟了一条新思路。
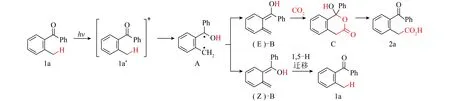
图6 邻烷基苯基酮与CO2的光催化羧化反应[33]Fig.6 Light-driven carboxylation of o-alkylphenyl ketones with CO2[33]
综上所述,光催化CO2耦合主要为CO2对C—O键和C—H键的插入。其中,对C—O键插入得到碳酸酯,由于C—O键极性高,易异裂,反应可以在温和的多相催化下进行,产率较高。而C—H键极性低且键能高,既不易异裂也不易均裂,因此C—H键的羧化反应需要在均相催化下才能顺利进行。此外,这些光催化反应大多在紫外光和紫光、蓝光下才能进行,占太阳光中大部分能量的长波段光还未有效利用,今后需进一步研究。
除活化反应分子外,促进反应物的吸附通常也有利于反应进行,因此将CO2捕集材料与催化剂复合,有望提高CO2转化的反应效率,国内外学者围绕(低浓度)CO2捕获封存开展了大量工作,有望为后期提升其光电转化催化耦合提供思路。近年来,北京林业大学王强教授团队[40-42]针对该领域进行深入研究,揭示了层状复合氢氧化物(水滑石、LDHs)材料具有优异的CO2捕获能力和对多种反应的催化性能。近期研究表明,LDHs具有优异的CO2催化转化性能[2,43],为设计和制备具有高效催化性能的LDH基催化剂提供了新思路。LDHs作为一种易大规模合成的材料,已经在山东万新威纳建立了年产3 000 t级的清洁工艺生产线,并成功应用于环境治理等方面[44]。未来LDHs在CO2耦合甚至其工业化应用中展现出巨大潜力。
2 电催化CO2耦合
系统论述电催化CO2耦合,围绕还原端有机化合物羧化反应,从有机分子活化出发,讨论该类反应的研究进展。该类反应的催化条件和效率见表3。
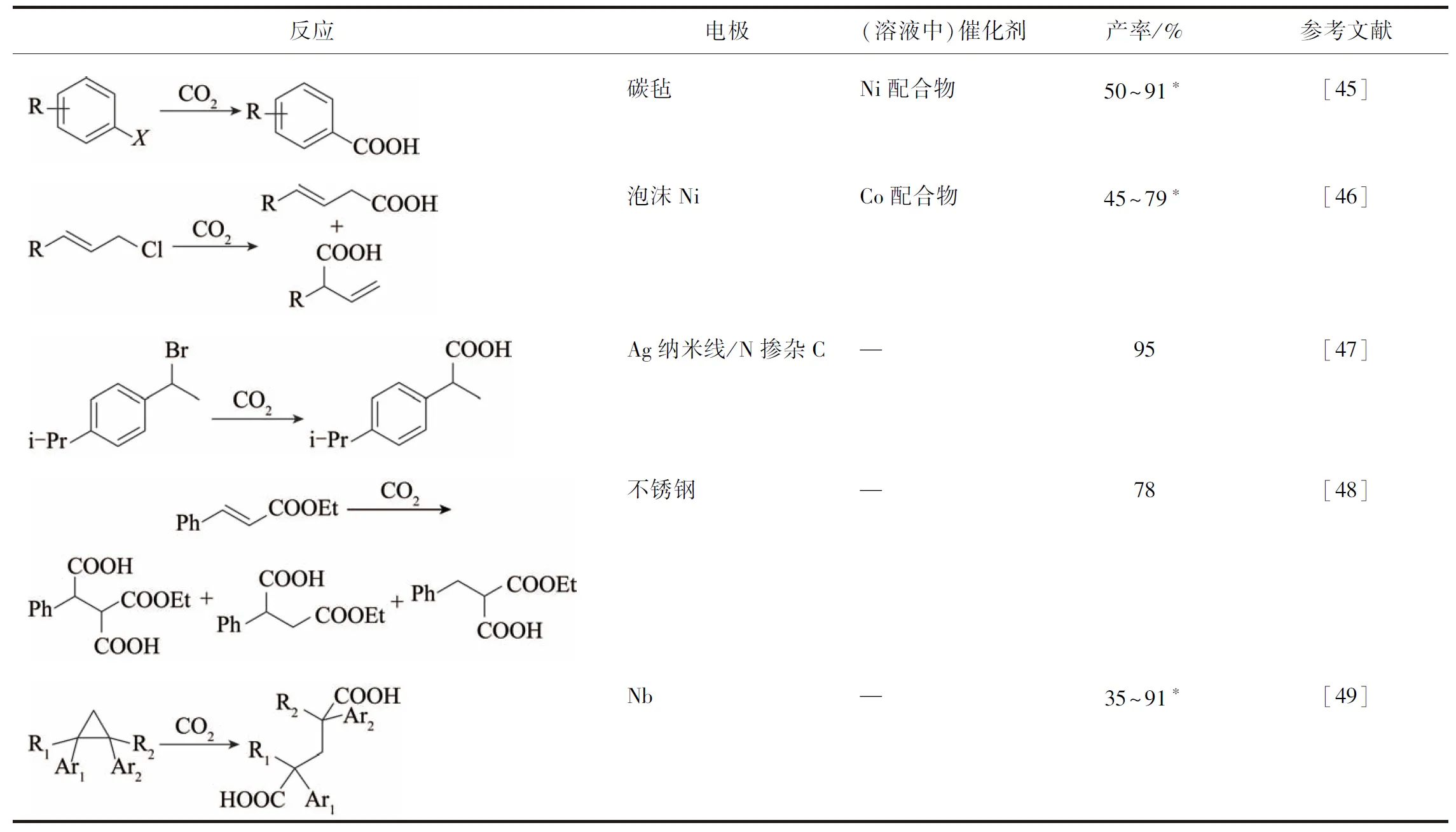
表3 CO2与有机物羧化中一些电催化反应的条件和效率Table 3 Conditions and efficiency of some electrocatalytic reactions in the carboxylation of CO2 and organics
2.1 C—X键的还原羧化
CO2与C—X(卤素)键的还原羧化生成对应的羧酸C—COOH,该反应可用于在有机分子的指定位置引入羧基。在经典的催化体系中,通常需加入过量金属有机试剂或金属单质作为还原剂促使反应发生[50-51],造成反应成本过高且整个反应不易控制。使用电还原策略进行C—X键的还原羧化能有效避免这些缺点,还可以减少能源或试剂的浪费。
与C—H键相比,C—X键键能更低、极性更强,通常更易于活化。低氧化态的过渡金属很容易与C—X键发生插入反应,这是很多过渡金属配合物催化有机反应的第一步。因此,沿用经典催化体系中的过渡金属催化剂是该电催化的策略之一,此时由阴极直接为催化循环提供电子,而非原来的金属颗粒。SUN等[45]报道了镍配合物催化下芳基卤化物与CO2于还原端生成羧酸的过程,反应机理如图7所示,具体包括卤代烃氧化加成、卤素离子消除、CO2插入和羧酸根离子消除,以及将穿插在其中的单电子还原。SOMERVILLE等[52]成功合成了Ni(I)中间体并研究了其与CO2的反应,证明了该反应机理的可靠性。不同过渡金属有不同的稳定氧化态,因此会在催化循环的不同位置获得电子[46],但反应机理一致。此外,SUN等[45]还开发了不牺牲阳极的方法,发现甲苯的电氧化氯化与之匹配良好,取得了理想效果。该方法为已有催化策略的改进,具有反应机理明确、产率和反应速率理想等优势,但催化剂以溶质形式存在于反应体系中,使得产物分离和催化剂循环利用较困难。
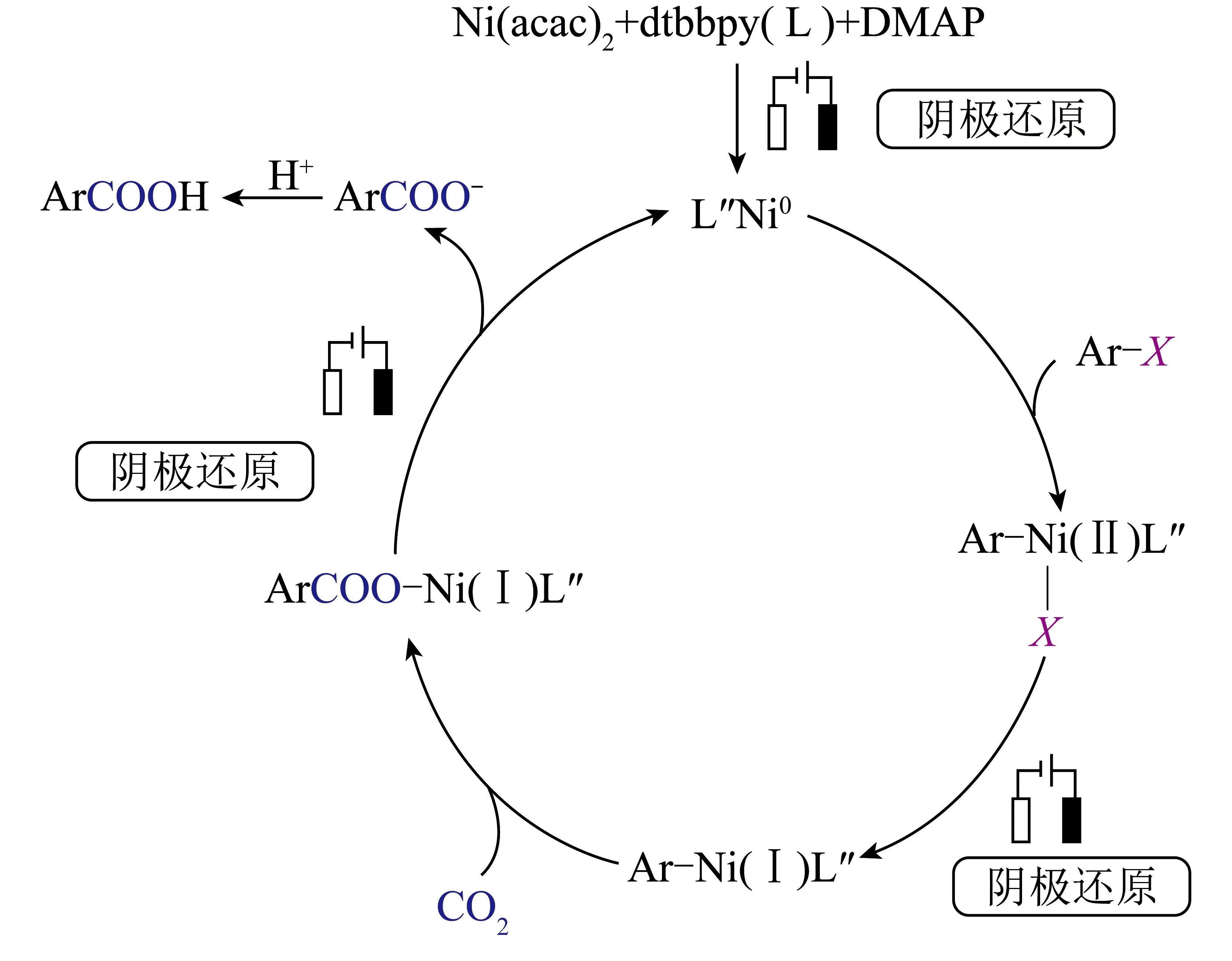
图7 镍催化的芳基卤化物与CO2的电化学羧化[45]Fig.7 Nickel-catalyzed electrochemical carboxylation of aryl halides with CO2[45]
已有研究表明,可通过改善阴极材料,将催化过程从液相转移至电极表面可解决这一问题,获得更高产率的羧酸。在电极表面,电子进入底物分子的LUMO,通常是C—Xσ*轨道,削弱其键级,促使化学键断裂形成卤素离子和自由基,通过自由基或碳负离子与CO2的偶联生成羧酸。YANG等[47]合成了具有核壳结构的银纳米线-氮掺杂碳复合材料,滴涂到电极上作为该反应的电催化。通过试验和理论计算,发现在银周围的氮掺杂碳壳可能会增加这些复合材料的电化学活性表面积,从而影响整体的电催化反应。虽然未对该反应机理作进一步研究,但指出了CO2吸附对该反应的影响。MEDVEDEV等[53]以银为阴极,将反应与电催化析氧(OER)耦合,避免了牺牲阳极,具有反应条件简单、反应成本低等优势。由于单质银电极无额外的催化效果,导致该反应体系产率较低,详细研究了反应过程和反应中各因素的影响,并提出了可能的反应机理,即电极表面碳负离子关键中间体的生成。不同电位范围下卤代烃和CO2电还原的可能机理和副反应如图8所示,E为电极电势(V vs Ag/Ag+),AdN为亲核加成反应,SN2为双分子亲核取代反应。反应需在适当的还原电位下进行以抑制副反应:电位过低时,CO2分子易被过度活化,增加了CO2还原反应的发生;电位过高时,R—X键断裂生成的自由基不足以被还原为碳负离子,导致其大量偶联为R—R。因此,如何改善电极材料以进一步提高反应速率和选择性,是今后研究重点。

图8 不同电位范围下卤代烃和CO2电还原的可能机理和副反应[53]Fig.8 Possible mechanism and side reactions of electroreduction of organic halides and CO2 in different potential ranges[53]
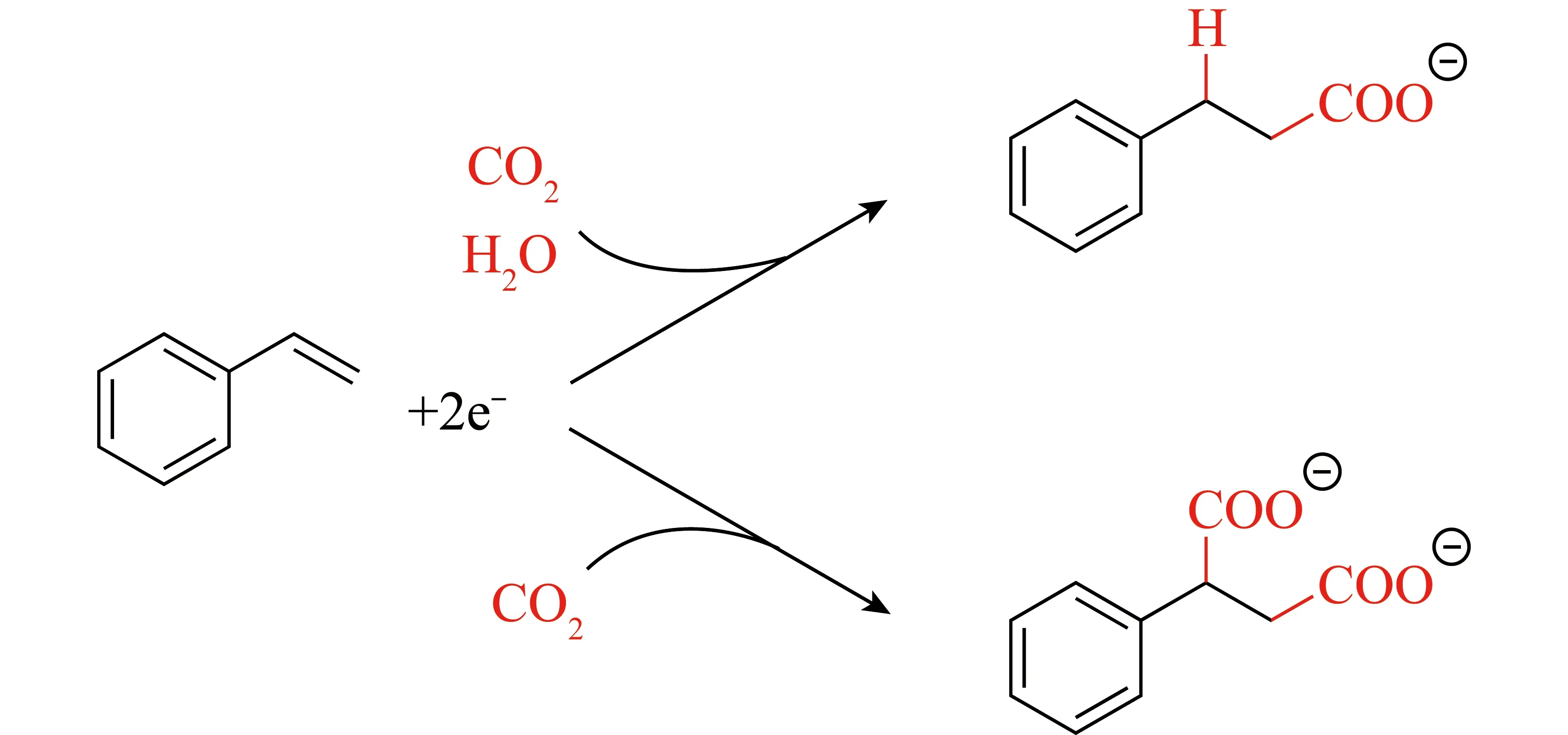
图9 苯乙烯电化学羧化[54]Fig.9 Electrochemical carboxylation of styrene[54]
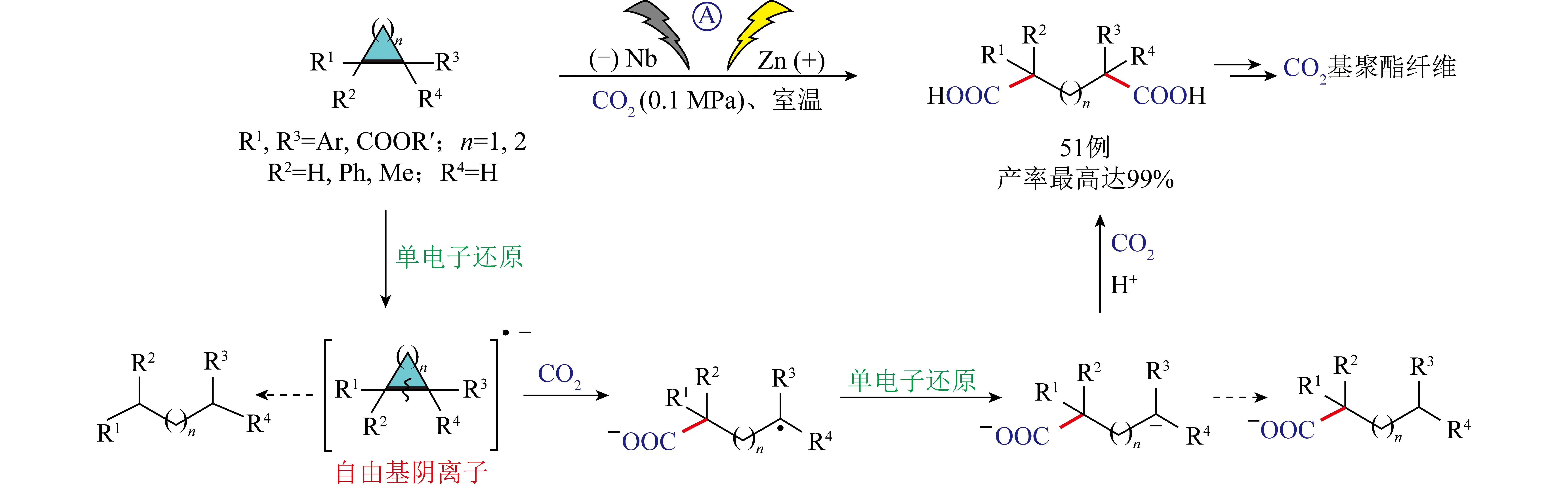
图10 C—C单键与CO2的电还原开环二羧化反应[49]Fig.10 Electro-reductive ring-opening dicarboxylation of C—C single bonds with CO2[49]
共轭二烯比单烯更易被还原,因而有更高的转化率,但其产物有更多可能性,因此,对共轭二烯电催化羧化的研究集中于提高某一种产物的选择性。早期研究通常在电催化体系中加入过渡金属,通过金属离子的络合控制反应选择性[55-56]。不使用均相催化剂时控制产物选择性成为挑战。SHETA等[57]发现不同电极材料对双键羧化和还原的选择性不同,相比Ni电极,使用不锈钢作为阴极可有效提高1-苯基-1,3-丁二烯在4号碳羧化的选择性,说明电极与其表面中间体之间的相互作用对反应有重要影响。
综上所述,电催化的CO2耦合转化率较高,可高效获得多种目标羧酸,且反应条件简单。然而大多反应选择性较差,仍使用牺牲阳极,开发高选择性的催化剂及利用氧化端反应是未来研究方向。
3 结语与展望
1)目前大多数光催化反应需在大功率光源照射下进行,才能取得理想的转化率和反应速率。降低光源功率或使用可见光时,反应转化率和速率可能大幅降低。因此,调控催化剂的微观结构,如电子结构、形貌、缺陷等,以增强对可见光乃至太阳光,特别是600 nm波长以上光的利用,有待进一步研究。
2)在多相催化领域中,具有高度可调变性的材料往往具有更好的催化效果。层状双金属氢氧化物(LDHs)是一类尺寸厚度、层间阴离子、层板元素以及形貌可调的二维纳米材料。目前,笔者课题组已利用该材料在CO2还原、水分解等多种反应中取得了较好的催化效果。随研究深入,LDHs有望成为CO2与有机化合物耦合反应的高效催化剂。
3)电催化反应中,大多研究主要关注位于阴极的CO2与耦合反应,而往往忽略对阳极的研究和应用。发展相应的电催化工艺,不仅要关注阴极的主要反应,还需考虑阳极的高效利用。
猜你喜欢
油气田地面工程(2022年8期)2022-10-02
陶瓷学报(2021年3期)2021-07-22
粉末冶金技术(2021年1期)2021-03-29
陶瓷学报(2020年6期)2021-01-26
陶瓷学报(2020年2期)2020-10-27
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
科技资讯(2018年16期)2018-10-26
分析化学(2018年12期)2018-01-22
科技资讯(2017年12期)2017-06-09
