
原发性骨内Rosai-Dorfman病的影像学表现及其病理相关性分析
2022-11-21 08:17刘世红胡美芳李红兵
中国CT和MRI杂志 2022年11期
刘世红 胡美芳 李 炜 夏 军 李红兵,*
1.深圳市宝安区福永人民医院放射科(广东 深圳 518103)
2.深圳市第二人民医院医学病理科(广东 深圳 518000)
3.深圳市第二人民医院医学影像科(广东 深圳 518000)
Rosai-Dorfman病是一种具有自限性的组织细胞增生性疾病,该病最早于1969年由Rosai 和 Dorfman提出并对其命名[1]。原发性骨内RDD定义为发生于骨且不伴淋巴结受累,至今较为少见,大部分为个案报道,2016年第五版WHO骨肿瘤分类将其归类于骨的造血系统肿瘤。作者对收集的4 例原发性骨内RDD病并结合国内外文献报道23个病例进行综合分析,以提高对该少见病的认识。
1 资料与方法
1.1 临床资料选取 2019年1月至2021年5月经手术病理证实的 4例患者的影像学资料同时进行文献复习27例,男性13例,女性14例,年龄2~72岁,平均年龄(33.55±19.38)岁。6例椎体及附件,5例股骨,4例胫骨,4例颅骨及颅底,3例桡骨,2例肱骨,1例骰骨,1例舟骨,1例肋骨,本组中4例均行手术切除。
1.2 检查方法4例RDD患者来自4家不同医院,采用不同的CT和MR设备,技术参数不同。27例中均行CT扫描,其中增强3例;13例行MR平扫,其中增强7例。CT和MRI增强扫描均采用动脉期、静脉期和延迟期扫描。所有原始数据经影像工作站进行数据后处理。
1.3 研究设计通过CHKD及PubMed检索主题词bone和Rosai-Dorfman,共检索文献69篇,经筛选共纳入文献17篇共23例骨内RDD病例,结合本组收集原发性骨内RDD患者4例合并分析,合计患者27例。
纳入标准:研究对象:通过病理诊断为骨内RDD患者;研究范围:关于RDD个案报道及论著;排除标准:无法获得文献全文者;重复报告者,包括同一作者在不同文章中报道的同一病例;文献综述等。
1.4 图像分析对27例骨内RDD在X线、CT、MRI及增强图像上病灶发生的部位、信号、边缘、骨质破坏程度、骨膜反应,是否突破骨皮质侵犯软组织及强化等特点进行分析,图像分析由两名具有20年以上 MR/CT 诊断经验的影像科医师进行共同探讨,达成一致。
2 结 果
2.1 影像学表现骨内RDD患者不同年龄均可发病,性别无明显差异,好发于长骨和不规则骨,27例中26例(占96.3%)影像学呈溶骨性破坏(图A~图D),仅有4例(占14.8%)边缘少许骨质硬化,3例(占11.1%)显示轻度骨膜反应,7例(占25.9%)可见软组织肿块,增强呈明显强化(图E)(附表1)。
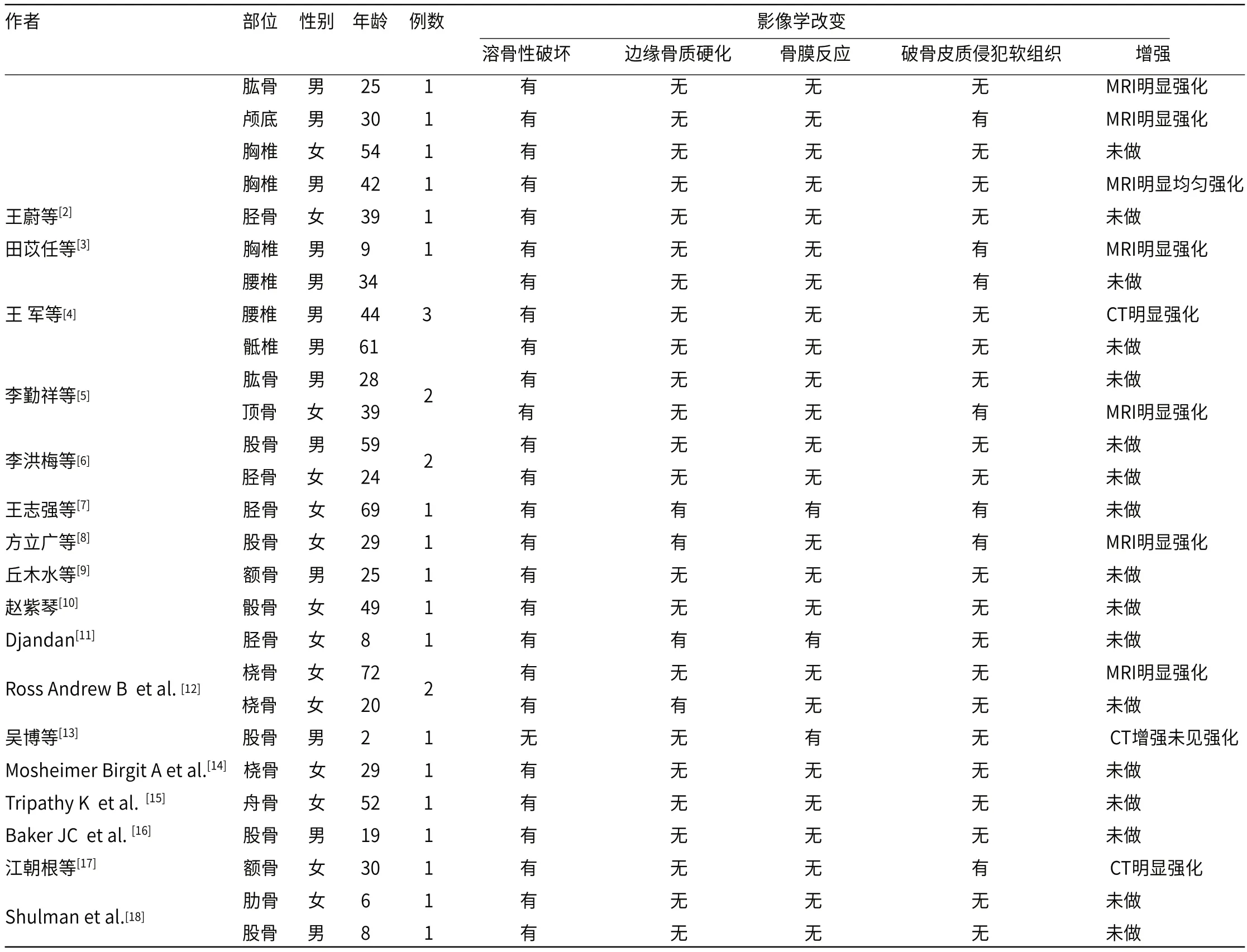
表1 27例骨内RDD影像表现
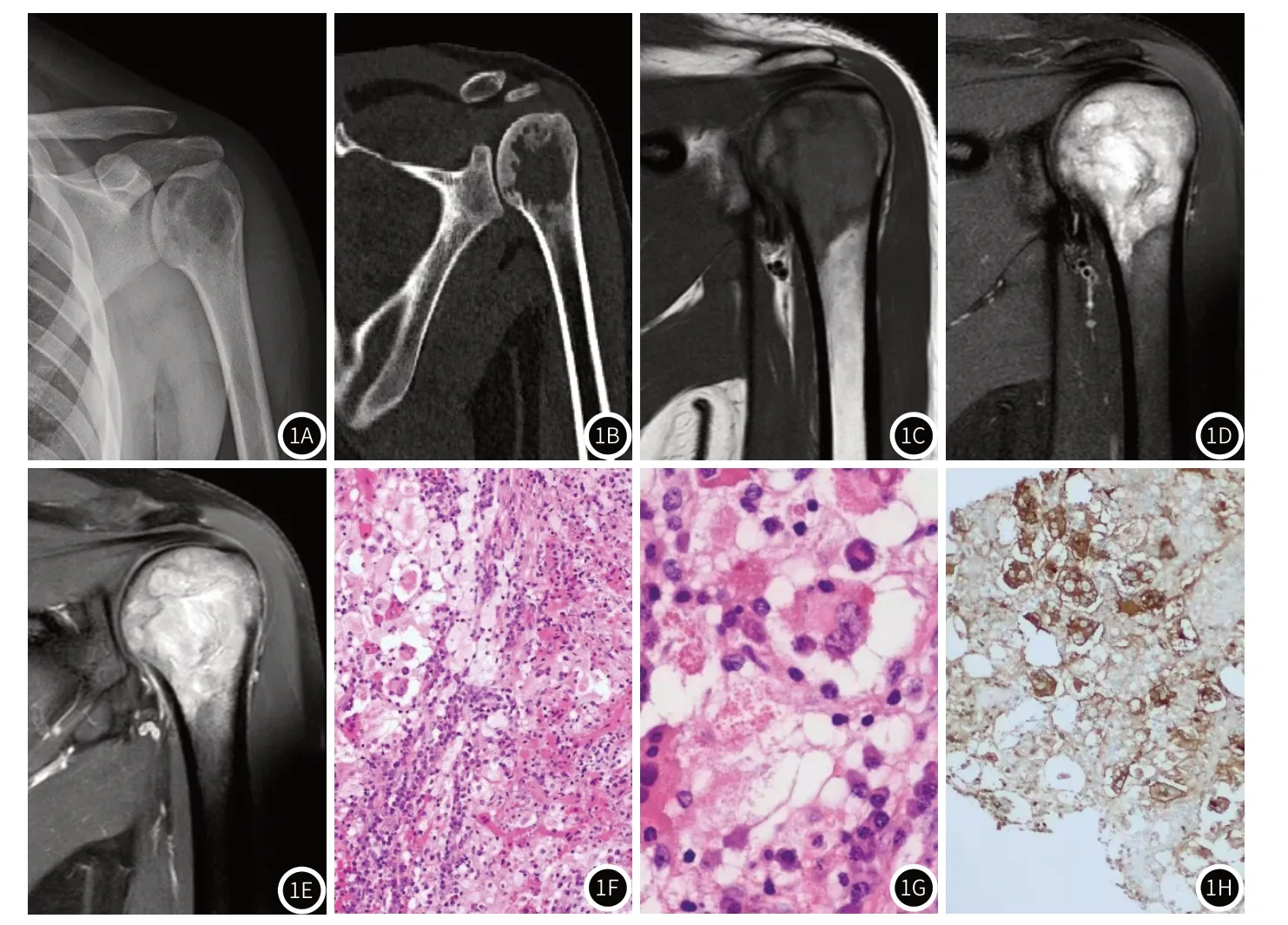
2.2 病理表现本文27例患者均可见明区的胞浆淡粉染的组织细胞和(或)泡沫样细胞及暗区的混杂淋巴细胞及浆细胞交替出现;局部可见吞噬形态完整的中性粒细胞、淋巴细胞和浆细胞等,纤维间质纤维化,免疫表型:S-100(+),CD68(+),CD1a(-)(图1F~图H)。
病例1,左肱骨RDD,图1A~图1E,A.X线肱骨溶骨性破坏,其上缘延伸至软骨下;B.CT示破坏区见残留骨嵴,未见骨膜反应;图1C~图1E依次为T1WI、T2WI压脂相及T1WI压脂增强图,显示周围软组织未见肿物,病灶区明显强化,图1F图为HE×100,镜下视野:可见明区、暗区交替出现,明区为胞浆淡粉染的组织细胞和(或)泡沫样细胞;暗区为混杂的淋巴细胞及浆细胞;局部可见吞噬现象,纤维间质纤维化,图1G图为HE×400,镜下视野:胞浆淡粉染的组织细胞内可见混入其中的淋巴细胞、浆细胞等炎症细胞,即吞噬现象(箭头标记所示),图1H图为IHC×200 S-100,显示病变中的组织细胞展现出的吞噬现象(即组织细胞内的淋巴细胞、浆细胞等炎症细胞无着色现象。箭头标记所示)s-100免疫标记为迈新生产,克隆号:4C4.9。
3 讨 论
3.1 骨内Rosai-Dorfman病概况组织细胞肿瘤是一种罕见的肿瘤,主要包括Erdheim-Chester病(ECD)、朗格汉斯细胞组织细胞增生症(LCH)和Rosai-Dorfman病(RDD),Meyo诊所报道[19],年发病率<1/200000人,一直以来,LCH及ECD被认为是炎症性疾病,最近有学者研究发现,LCH及ECD患者丝裂原活化蛋白激酶/细胞外信号调节激酶(MAPK/ ERK)通路的突变,对它们是肿瘤性病变提供了有力的证据支持[20],另外33%的RDD患者中也报道了类似突变。这导致它们在2016年世界卫生组织被列入造血和淋巴系统肿瘤[21]。
3.2 骨内Rosai-Dorfman病病理组织学和影像学表现
3.2.1 病理组织学 原发性骨内RDD文献报导[22]极其少见,骨内RDD的病理组织学特点有[2]:具备吞噬能力的组织细胞数量相对减少,导致吞噬不明显,故镜下很难被发现,而组织细胞增生显著,常呈梭形改变,编织状、席纹状排列,周围可见少许浆细胞及淋巴细胞浸润,间质纤维化反应更加明显,不同病例增生的组织细胞类型不同(图1F~图1H),本文21例均发现不同组织细胞增生及周围细胞浸润与文献相符;骨内RDD病较易向周围侵袭性生长,侵犯并破坏骨及软骨组织,本文病例均表现溶骨性破坏,与文献报道其侵袭性生长的病理特点符合。除了细胞形态学的改变,免疫组织化学 CD1a(-)、CD68(+)、S-100(+)是主要依据,其中S-100(+),CD1a (-),表达部分单核-巨噬细胞;标志物如CD68:说明这种细胞是一种巨噬细胞与树突细胞的杂交表型,本文骨内RDD免疫表现均符合上述改变。
3.2.2 影像学表现 骨内RDD是极其少见的且容易误诊为炎症或其他肿瘤性病变,本组27例发病年龄范围为2-72岁,平均年龄(33.55±19.38)岁,与文献报道基本符合;根据发病部位:本组27个病例发生于长管状骨13例,占本组48%,颅骨4例(14%),脊柱6例(22%),不规则骨3例(11%),与既往研究报道常见于长管状骨、颅骨及脊柱[23]相符;本组27个病例中有26例骨内RDD在X线及CT上均表现为不规则溶骨性骨质破坏,其中23例边缘未见骨质硬化,1例见残留骨嵴,MRI上多表现为T1WI为低信号,T2WI为稍高信号影,周围未见明显水肿,与王蔚等研究发现骨内RDD类似,残留骨嵴说明该病呈慢性生长,而病灶溶骨性破坏及边缘未见骨质硬化说明该病具有一定的侵袭性,一般不突破骨皮质[2-18]。本组27个病例中24例未见明显骨膜反应,但有3例出现骨膜反应,7例突破骨皮质累及软组织。王志强等报道3例骨内RDD周围可见骨膜反应,并累及邻近软组织,说明病灶一旦突破骨皮质,有可能引起相应骨膜反应及软组织肿胀,骨膜反应可能为大量组织细胞破坏骨皮质所致,软组织肿胀可能为梭形的组织细胞和增生的胶原[7,11,13];本文27个病例,其中10例行增强扫描,9例显示明显强化,说明该病具有比较丰富的血管,然这一征象有待于更多的病例证实;另外吴博报道股骨病灶1例[13],可见明显骨膜反应,未见骨质破坏及侵犯周围软组织,此为个例,既往无文献报道,可能与病程较短有关,因无MRI检查,故骨内早期病变侵犯范围不能很好显示。
3.3 骨内Rosai-Dorfman病鉴别诊断骨内RDD表现为溶骨性破坏,边缘骨质硬化少见,骨膜反应及明显软组织肿块更加少见,需要与以下疾病鉴别:①感染性病变:起病急,临床症状明显,早期骨质破坏,骨膜反应明显,中晚期骨质增生硬化,骨膜反应消失或减轻。②骨嗜酸性肉芽肿[24]:男性,儿童多见,溶骨性骨质破坏,骨膜反应少见,未见软组织肿块,周围软组织水肿明显,免疫组化CD1a为阳性作为主要鉴别手段。③淋巴瘤[25]:发病年龄比较大,临床症状相对轻微,虫蚀状骨质破坏,软组织肿块明显,肉包骨影像改变,骨膜反应一般较轻,增强扫描有不同程度强化。④浆细胞瘤:溶骨性骨质破坏,轻度膨胀性,椎体常见脑回征,周围有软组织肿块。⑤转移瘤:一般病例都有原发病灶,年龄较大,以多发为主。
综上所述,骨内RDD患者不同年龄均可发病,影像上呈溶骨性骨质破坏,骨膜反应及边缘骨质硬化少见,部分可突破骨皮质侵犯周围软组织,增强明显强化,软组织肿块更加少见,部分与病理有一定相关性,该病的确诊主要依靠细胞形态学及免疫组织化学检查。
猜你喜欢
首都食品与医药(2022年12期)2022-12-06
中国临床医学(2022年3期)2022-07-08
临床超声医学杂志(2022年4期)2022-05-07
粘接(2022年3期)2022-04-19
医学信息(2021年5期)2021-03-21
影像研究与医学应用(2021年2期)2021-03-09
发明与创新·中学生(2018年11期)2018-11-30
特别健康·下半月(2017年7期)2017-08-09
安徽医科大学学报(2015年9期)2015-12-16
医学研究生学报(2012年8期)2012-12-25
