
COX20基因变异线粒体复合物Ⅳ缺乏症相关的遗传性周围神经病4例病例系列报告及文献复习
2022-11-29 06:36胡超平施亿贇李西华周水珍
中国循证儿科杂志 2022年5期
胡超平 施亿贇 李西华 赵 蕾 周水珍 王 艺
线粒体病是一组具有高度临床和遗传异质性的遗传性代谢疾病,由线粒体基因(mtDNA)或核基因(nDNA)变异导致,多系统受累是其常见的临床表现。神经元的生长、存活和功能都与线粒体息息相关,线粒体产生的ATP为突触的组装、动作电位的形成及突触递质的传递等提供能量[1],因此线粒体病患者常伴随周围神经系统受累[2]。然而,大部分线粒体病相关周围神经病均以亚临床改变为主,以周围神经系统症状为主要或突出表现的线粒体病目前国内外报道较少,主要见于MFN2和GDAP1基因变异患者[3, 4],两者也是遗传性周围神经病的重要病因。2013年,有学者首次报道1例COX20基因(即FAM36A基因)变异的患者线粒体复合物Ⅳ水平下降,功能受损,表现为共济失调和肌张力低下[5],此后多个中心相继报道了COX20基因变异引起的周围神经病、小脑共济失调等病例[6-9],扩展了线粒体病及遗传性周围神经病的基因变异谱系。本文收集了复旦大学附属儿科医院(我院)近年诊治的COX20基因变异的线粒体病连续病例并行文献复习,总结其临床表现和基因型特征及随访、预后情况,以期提高临床医师对该病的认识。
1 病例报告
例1,女,足月顺产,无窒息抢救史。自幼精神运动发育落后于同龄儿(发育里程不详),进食容易呕吐。独走后一直步态异常,5~6岁后出现倒退,表现为行走不稳、口齿不清、反应迟钝,10岁左右丧失独立行走能力。14岁至我院门诊就诊,查体:神清,精神反应可,理解能力差,构音不清,颅神经未见异常,双上肢肌力MRC评分Ⅴ级,双下肢肌力Ⅲ+级,双侧膝反射未引出,巴氏征可疑阳性,下肢马蹄内翻足畸形。心肌酶谱和乳酸均在正常范围,尿气相质谱提示柠檬酸明显升高、乌头酸略高,血串联质谱未见异常。头颅MR提示脑沟略明显,头颅磁共振波谱成像未见异常代谢波谱。心电图提示窦性心律不齐。肌电图提示多发性周围神经损害,主要累及感觉神经,运动神经亦有累及,轴索病变为主(表1)。不规则服用维生素B类药物,16岁末次随访时,下肢严重畸形,上肢肌力下降不明显,但出现手抖、握物不稳。
例2,例1胞弟,孕期及出生史未见异常。18月龄独走,智力低下,4~5岁出现踮脚走路,之后下肢无力缓慢进展,9岁因“运动功能明显退步”至我院门诊就诊,查体:行走鸭步,口齿欠清。四肢远端肌张力偏低,膝腱反射未引出,双足弓偏高,双上肢肌力正常,双下肢肌力Ⅲ级。心肌酶谱正常范围,肌电图发现多发性周围神经损害表现,主要累及感觉神经,运动神经亦有累及,以轴索病变为主(表1)。予口服维生素B12,患儿病情仍逐渐进展。10岁不能独走;11岁出现马蹄足畸形;13岁末次随访时,下肢畸形加重,上肢功能无明显受累,仍口齿不清,智力倒退不明显。
例3,女,出生史未见异常。5月龄抬头,9月龄独坐,20月龄独走。4岁5月龄仍步态不稳,不会爬梯、跑跳。语言发育未见异常。平日运动耐受差,劳累及感冒后症状加重,休息及急性感染恢复后运动耐受能力可恢复到感染前的基线水平。至我院门诊就诊,查体:神清,精神反应可,四肢肌张力低下,腱反射未引出,肌力基本正常,病理征(-)。下蹲起立慢,不会跳。CK在正常范围。肌电图提示多发性周围神经损害,累及感觉神经,运动神经稍累及,部分肌肉合并可疑肌源性损害,肱二头肌为主(表1)。尿气相质谱提示,3-羟基丁酸、乙酰乙酸增高,辛二酸、己二酸略高,血串联质谱未见明显异常。予口服艾地苯醌等治疗及康复训练后,短时间内运动能力略有改善,能跳。后期不规则康复,行走能力较前退步。7岁6月龄末次随访时,肌力无明显下降,行走明显不稳,上下楼费力,协调能力较前稍差,偶有口齿不清,认知无倒退。
例4,男,35+4周早产出生(其母妊娠期糖尿病及高血压病史),出生体重2 200 g,生后发现远视(200~300度)。16月龄独走,但一直行走不稳,平衡能力欠佳,上下楼费力,语言发育未见异常。5岁时因“行走不稳,姿势异常”至我院门诊就诊,查体:神清,精神反应好,颅神经未见异常。四肢肌张力偏低,膝腱反射未引出,肌力未见异常。血串联质谱及尿气相质谱未见异常,血磷酸肌酸激酶 292(正常参考值<210)IU·L-1,乳酸正常,头颅MR未见异常。肌电图提示多发性周围神经损害,累及感觉和运动神经,轴索病变为主(表1)。予以鸡尾酒疗法。目前7.5岁,行走不稳同前,逐渐出现口齿欠清和反应稍迟钝。
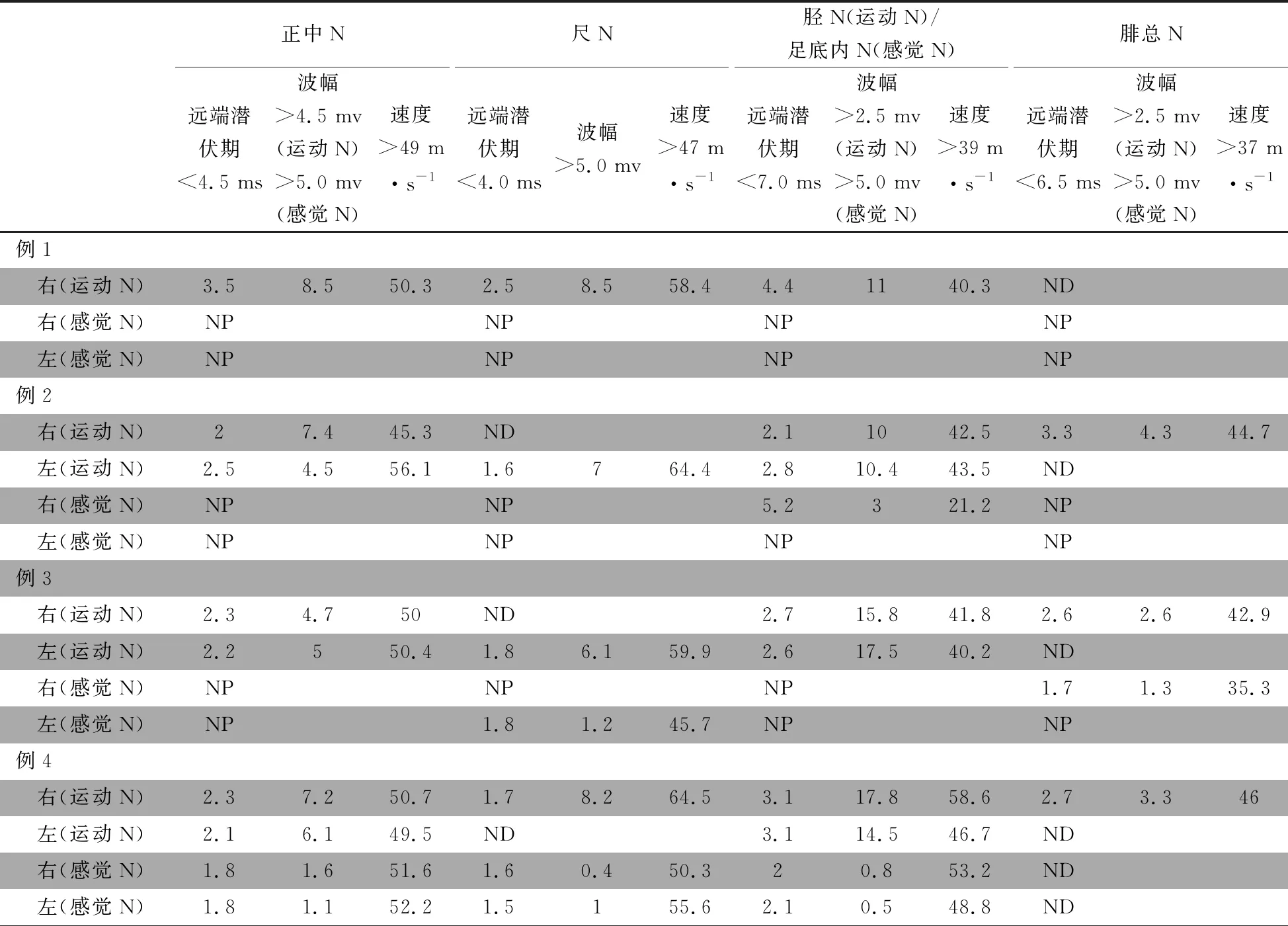
表1 4例COX20基因变异患儿的神经传导检查结果
取得患儿父母的知情同意后,采集家系外周静脉血。采用 miniblood全血试剂盒(QIAGEN 公司,德国)提取基因组DNA, NanoDroop 2000(Thermofisher公司,美国)紫外分光光度仪测定其浓度。基于Illumina Hiseq 2000/2500平台进行序列检测。测序覆盖度>98%,平均测序深度>100×。采用美国医学遗传学与基因组学学会(ACMG)发布的《序列变异解读标准和指南》进行分类[10]。4例患儿全外显子测序结果均发现存在COX20基因复合杂合变异(图1A)。例1和2存在无义变异c.259C>T(P.Q87X)和错义变异c.41A>G(p.K14R),分别来自于母亲和父亲,均为HGMD已报道的致病变异。例3存在2个错义变异:c.41A>G(p.K14R)和c.98C>T(p.S33L),分别来自于父亲和母亲,均为已报道的致病变异。例4存在移码变异c.262delG(p.E88Kfs*35)和错义变异c.41A>G(p.K14R),分别来自于母亲和父亲,其中移码变异尚未见报道,位于4号外显子,262位单个碱基的缺失造成后续密码子的框移改变,从而影响蛋白功能,根据ACMG分类,此变异为致病性(PVS1+PM2+PM3+PP4)。
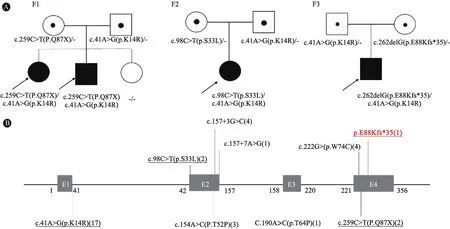
图1 4例COX20基因变异患儿的家系图及目前报道的COX20基因致病变异位点在DNA水平的分布图
以“COX20”、“线粒体复合物Ⅳ缺乏症”为关键词在万方数据知识服务平台、中国期刊全文数据库(CNKI)中检索中文文献;以“COX20”、“complex Ⅳ deficiency”为关键词在生物医学文献数据库(PubMed)和美国国家生物技术中心(NCBI)检索英文文献。检索时间为建库至2021年12月。中文数据库未检索到目标文献,在PubMed和NCBI数据库分别检索到10篇和9篇目标文献,排除基础研究12篇和重复发表1篇,余6篇文献和本文共报道22例COX20基因变异的患儿来自18个家系(表2)。1例宫内发育迟滞,2例生后肌张力低下,11例精神运动发育落后。22例患儿首发症状均为行走困难或步态不稳,起病中位年龄5(1.0~17)岁。病程中14例(63.6%)出现构音障碍,中位年龄6(2~19)岁;14例(63.6%)出现肌力下降和/或足部畸形,中位年龄12.5(7~29)岁;8例(36.4%)出现共济失调,中位年龄5(1.4~16)岁;6例(27.3%)出现肌张力障碍,中位年龄11.5(5~19)岁;5例(22.7%)存在认知倒退,中位年龄7(5~18)岁;3例(13.6%)存在抑郁倾向;2例(9.1%)合并注意力缺陷多动障碍;2例(9.1%)有眼外肌麻痹;1例(4.5%)存在吞咽障碍。9例(40.9%)已无法独立行走,丧失独走能力的中位年龄为10(7~21)岁。21例患儿行神经传导及肌电图检查,19例(90.5%)提示多发性周围神经病变,其中14例为感觉和运动神经均受累,5例以感觉神经受累为主。头颅MR检查18例,小脑萎缩4例(22.2%);脊髓MR检查10例,脊髓萎缩4例(40%)。

注 病例1和2、病例15 和16、病例18和19、病例21和22 为同胞。ADHD:注意力缺陷多动障碍。S:感觉神经轴索损害,M:运动神经轴索损害;NA:数据无法获得或未做
图1B显示,22例患儿均发现COX20基因变异,7例为纯合变异,15例为复合杂合变异。共发现9个变异位点,其中错义变异5个:p.K14R(n=17)、p.W74C (n=4)、p.T52P (n=3)、p.S33L(n=2)、p.T64P (n=1);剪切变异2个:c.157+3G>C (n=4)、c.157+7A>G(n=1);无义变异1个:p.Q87X (n=2);移码变异1个:p.E88Kfs*35 (n=1)。
3 讨论
细胞色素C氧化酶(COX)缺乏症除少数由编码亚基的基因变异导致[11]以外,多由组装因子如SURF、COA5等基因变异引起,具有高度的临床异质性,以线粒体肌病、心肌病、Leigh综合征等为主[12, 13],大多存活率低[14],周围神经病变报道则较少[15, 16]。本文报道的4例COX20基因变异的线粒体病患儿,丰富了线粒体病的临床表型谱,扩大了遗传性周围神经病的疾病谱系。同时本文报道了COX20基因1个新的未报道的移码变异c.262delG(p.E88Kfs*35),扩展了COX20基因的变异谱。
总结目前国内外报道的22例COX20基因变异患儿的临床表现,总体呈现为早期起病、慢性进展的病程,核心症状主要包括轴索性周围神经病、共济失调、肌张力障碍、构音障碍、认知障碍等。大部分患者自幼运动发育里程落后,部分伴智力低下。多在儿童期出现步态不稳、平衡力差,同时或随后逐渐出现共济失调、构音障碍,而后进一步出现肌力下降及足部骨骼畸形、肌张力障碍、认知倒退等。随着病情进展,患者逐渐丧失独走能力,部分患儿晚期可伴随上肢及自主神经功能障碍,严重影响生活质量和日常功能。总之,患儿表现为进行性加重的过程,部分病例(如本文例3)病程中存在运动能力波动及运动不耐受的现象,行走不稳等症状在急性感染后加重,感染控制后短暂恢复至原有水平,这与多种其他线粒体病患者感染诱发或加重病情的临床表现相似,可能与线粒体病在急性病期的能量危象有关。
在COX20基因变异的患者众多临床表现中,轴索性周围神经病变,尤其是感觉神经受累最为突出,呈现出非长度依赖性的轴索型感觉神经病变特征,区别于一般的遗传性周围神经病表现。周围神经受累在线粒体病中并不少见,如POLG等基因变异导致的线粒体病相关周围神经病[2],MFN2、COX6A1、GDAP1及SCO2等基因变异导致的遗传性周围神经病等[3, 4, 16]。COX20基因变异相关周围神经病,以感觉神经受累最为显著,可能与组织特异性即在本体感觉神经元中的优先表达有关[9]。有学者提出COX20基因变异是常染色体隐性遗传的遗传性感觉神经病的潜在病因之一[9],临床工作中对于不明原因的多发性周围神经病,尤其是非长度依赖的感觉神经轴索病变,需考虑排查COX20基因变异等线粒体病的可能性。COX20基因变异患者出现的构音障碍和眼外肌麻痹等,提示舌咽、舌下及动眼神经等颅神经受累,可能是周围神经病变表现的一部分,但需要更多的研究证据支持。
构音障碍、共济失调和肌张力障碍也是COX20基因变异的常见症状,且构音障碍及共济失调在疾病早期即可出现。部分COX20基因变异的患儿头颅MR提示存在小脑萎缩。小脑性共济失调和肌张力障碍常见于一些神经变性病,如脊髓小脑性共济失调、Friedreich共济失调、Wilson病等[17]。本文病例提示,对于构音障碍、共济失调、肌张力障碍合并周围神经病变的患者,也需要考虑COX20基因变异等代谢性疾病的可能。

目前文献报道的COX20基因变异位点中,错义变异最常见,其次为剪切变异、无义变异和移码变异。然而,多个错义变异在改变氨基酸的同时,也可影响剪切。如1号外显子的错义变异c.41A>G(p.K14R)导致赖氨酸变异为精氨酸,又因其靠近内含子-1 (IVS1)的剪接供体位点,同时引起了该位点的剪接改变。c.154A(p.Thr52Pro) 位于2号外显子倒数第4个碱基,不仅导致高度保守的苏氨酸变异为脯氨酸,且造成2号外显子的剪切异常,生成截短的不完整蛋白(△EXON2),从而影响其功能[5]。在所有变异位点中,c.41A>G(p.K14R)占目前报道所有变异位点的48.6%,是COX20基因的热点变异位点。该变异在国内人群中的比例(0.001 32)远高于东亚人群(0.000 035 52),国内学者通过8个家系的单倍体分析证实此变异为中国东部人群的奠基者变异[9]。
本病目前缺乏特异有效的治疗方法,以对症支持和能量补充为主,但总体而言效果欠佳,行走困难、认知下降等呈逐渐进展的过程。大多数患儿在10岁左右丧失行走能力,致残率高。临床表型和基因型关系尚不明确,遗传背景或表观遗传等其他因素是否在表型的调控中发挥作用有待进一步阐明。
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
家庭医药(2022年2期)2022-02-18
全科护理(2022年3期)2022-02-18
中国康复理论与实践(2020年1期)2020-03-28
中南医学科学杂志(2019年6期)2019-12-05
中国民间疗法(2017年11期)2017-12-18
中国现代神经疾病杂志(2017年1期)2017-03-29
广东药科大学学报(2016年6期)2016-03-10
中国神经免疫学和神经病学杂志(2014年5期)2014-05-08
