
一测多评法同时测定连翘归尾颗粒中3种成分
2022-12-05 10:48遆安航李思思翟康欣谭丽媛张淑蓉
中成药 2022年9期
遆安航, 李思思, 陈 瑾, 翟康欣, 谭丽媛, 张淑蓉
(山西中医药大学,山西 晋中 030600)
连翘归尾煎出自《景岳全书》,由连翘、归尾、甘草、金银花、红藤组成,功效清热解毒、活血消肿[1],可治疗乳痈、口疽、颊疡、牙痈、猪丹毒等[2],方中连翘、金银花为君药,具有清热解毒、散结消肿作用[3]。课题组前期发现,该方在体内外实验中均显示对乳腺癌细胞的抑制作用,为了更有效地开展后期研究,保证实验样本前后的一致性,已将其改制成颗粒剂[4]。
为了降低检测成本,同时结合中药中多种成分相互作用、复杂多变等特点[5],采用一测多评法测定多指标成分含量已成为发展趋势和研究热点[6]。因此,本实验建立该方法同时测定连翘归尾颗粒中连翘酯苷A、绿原酸、连翘苷的含量[7],以期为该制剂质量控制和评价提供参考。
1 材料
1.1 仪器 数显电热套购自金坛市杰瑞尔电器有限公司(型号HDM500-10450);中药材粉碎机购自广州真麦机械设备有限公司(型号LG-50);恒温水浴锅购自上海比朗仪器制造有限公司(型号HH-2);旋转蒸发仪购自巩义市英峪弘源仪器厂(型号RE-5205);超声波清洗机购自杭州法兰特超声波科技有限公司(型号SB-5200 DTDN);高效液相色谱仪购自杭州瑞析科技有限公司(型号Waters e2695)、上海诺析仪器有限公司(型号Agilent 1260);电动喷雾器购自武汉药科新技术开发有限公司(型号CS-1020E);电子天平(十万分之一)购自瑞士梅特勒-托利多公司(型号AE240)。
1.2 试剂与药物 连翘归尾颗粒(批号201020、201121、201222)及各阴性颗粒均为自制。绿原酸(批号110753-201716,纯度>98%)、连翘酯苷A(批号111810-201606,纯度>98%)、连翘苷(批号110821-200610,纯度>98%)对照品及连翘(批号120908-201216)、金银花(批号121060-201608)、甘草(批号120904-201605)、大血藤(批号121353-201704)对照药材均购自中国食品药品检定研究院。乙腈为色谱纯(河南萨默生物科技有限公司);甲酸、甲醇为分析纯;水为娃哈哈纯净水。
2 方法与结果
2.1 色谱条件 Thermo Scientific BDS HYPERSIL C18色谱柱(150 mm×4.6 mm,5 μm);流动相乙腈(A)-0.1%甲酸(B),梯度洗脱(0~6 min,5%~9%A;6~9 min,9%~12%A;9~22 min,12%~14%A;22~35 min,14%~24%A;35~40 min,24%~26%A);体积流量1.0 mL/min;柱温25 ℃;检测波长278 nm;进样量10 μL。
2.2 溶液制备
2.2.1 供试品溶液 精密称取本品粉末(过6号筛)2 g,置于具塞锥形瓶中,加20%乙醇10 mL,密塞,浸泡40 min,称定质量,超声(功率250 W、频率45 kHz)处理45 min,放冷,20%乙醇补足减失的质量,摇匀,滤过,0.45 μm微孔滤膜过滤,即得。
2.2.2 对照品溶液 精密称取连翘酯苷A、连翘苷、绿原酸对照品适量,置于5 mL量瓶中,50%甲醇定容至刻度,摇匀,即得(三者质量浓度分别为0.538、0.066、0.222 mg/mL),在4 ℃下保存。
2.2.3 阴性样品溶液 将缺连翘、缺金银花和大血藤阴性样品研成粉末,过6号筛,分别精密称取2 g,按“2.1.2”项下方法制备,即得。
2.3 专属性考察 取“2.2”项下供试品、对照品、阴性样品溶液适量,在“2.1”项色谱条件下进样测定,结果见图1。由此可知,各成分色谱峰均能达到基线分离,表明该方法专属性良好。
2.4 线性关系考察 分别精密量取“2.2.2”项下对照品溶液4、8、16、24、32、40 μL,置于2 mL量瓶中,50%甲醇定容至刻度,摇匀,得不同质量浓度,在“2.1”项色谱条件下进样测定。以对照品峰面积为纵坐标(Y),质量浓度为横坐标(X)进行回归,得方程分别为连翘酯苷AY=888 061X-51 410(r=0.999 9),线性范围1.08~10.76 μg/mL;连翘苷Y=747 692X-3 710(r=1.000 0),线性范围0.13~1.32 μg/mL;绿原酸Y=1 798 549X-26 262(r=0.999 9),线性范围0.44~4.44 μg/mL,均呈现良好的线性关系。
2.5 精密度试验 精密量取“2.2.2”项下对照品溶液16 μL,置于2 mL量瓶中,50%甲醇定容至刻度,摇匀,在“2.1”项色谱条件下进样测定6次,测得连翘酯苷A、连翘苷、绿原酸峰面积RSD分别为0.16%、0.07%、0.48%,表明仪器精密度良好。
2.6 重复性试验 将同一批本品(批号201020)研成粉末,过6号筛,精密称取6份,每份2 g,按“2.2.1”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,测得连翘酯苷A、连翘苷、绿原酸含量RSD分别为2.88%、2.22%、2.65%,表明该方法重复性良好。
2.7 稳定性试验 将本品(批号201020)研成粉末,过6号筛,精密称取2 g,按“2.2.1”项下方法制备供试品溶液,置于4 ℃冰箱中保存,每隔4 h取出,在“2.1”项色谱条件下进样测定,测得连翘酯苷A、连翘苷、绿原酸峰面积RSD分别为0.58%、1.03%、2.47%,表明溶液在24 h内稳定性良好。
2.8 加样回收率试验 将各成分含量已知的本品(批号201020)研成粉末,过6号筛,精密称取1 g,置于锥形瓶中,精密加入适量对照品溶液,平行9份,按“2.2.1”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,计算回收率[8],结果见表1。
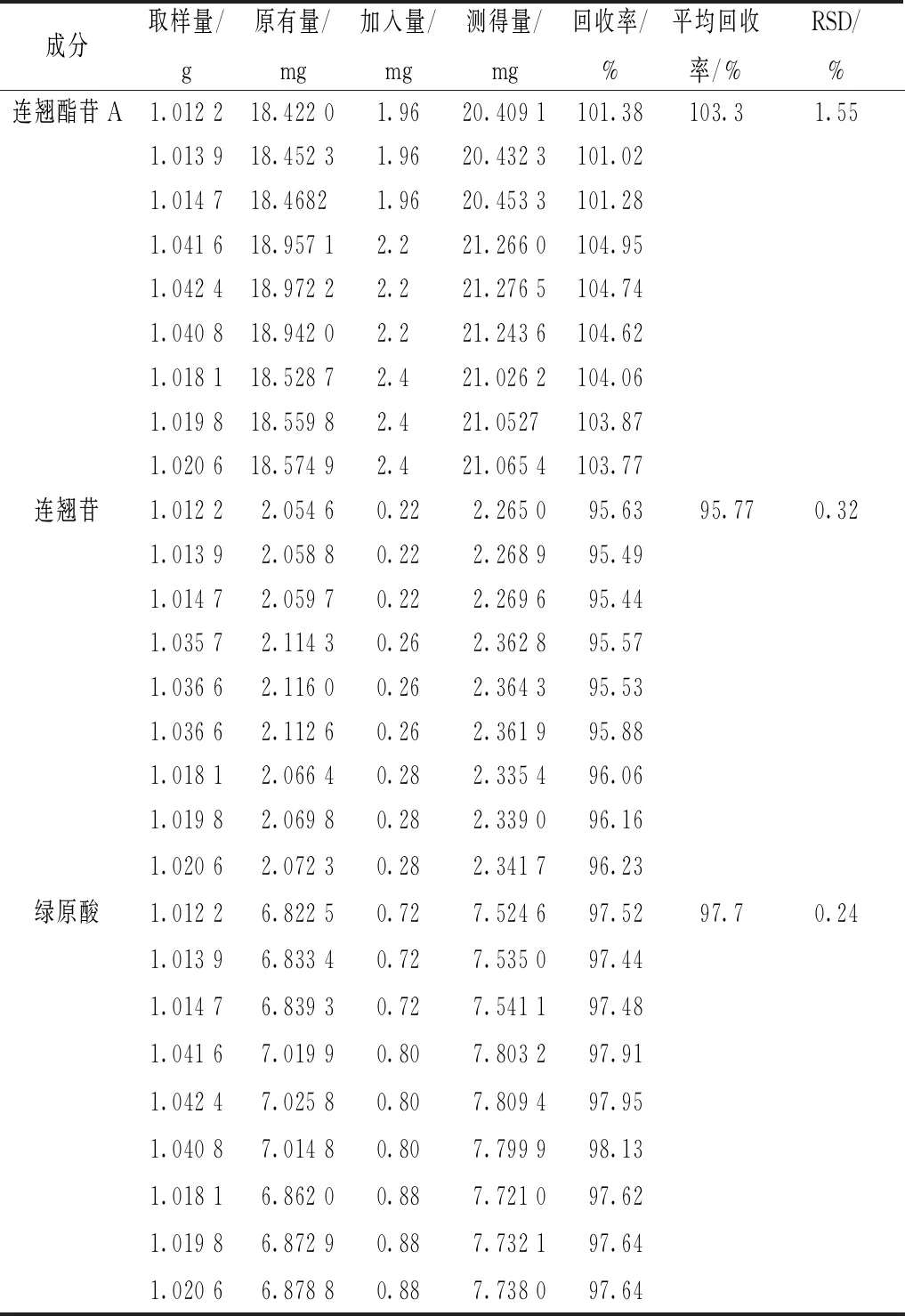
表1 各成分加样回收率试验结果(n=9)
2.9 相对校正因子计算 精密量取“2.2.2”项下对照品溶液5、10、15、20、30 μL,置于2 mL量瓶中,50%甲醇定容至刻度,摇匀,得不同质量浓度,在“2.1”项色谱条件下进样测定,以连翘酯苷A为内标,计算其他2种成分相对校正因子fk/s[9],公式为fk/s=fk/fs=(CkAs)/(CsAk),其中Ck为其他成分含量,Ak为其他成分峰面积,Cs为内标含量,As为内标峰面积,结果见表2。
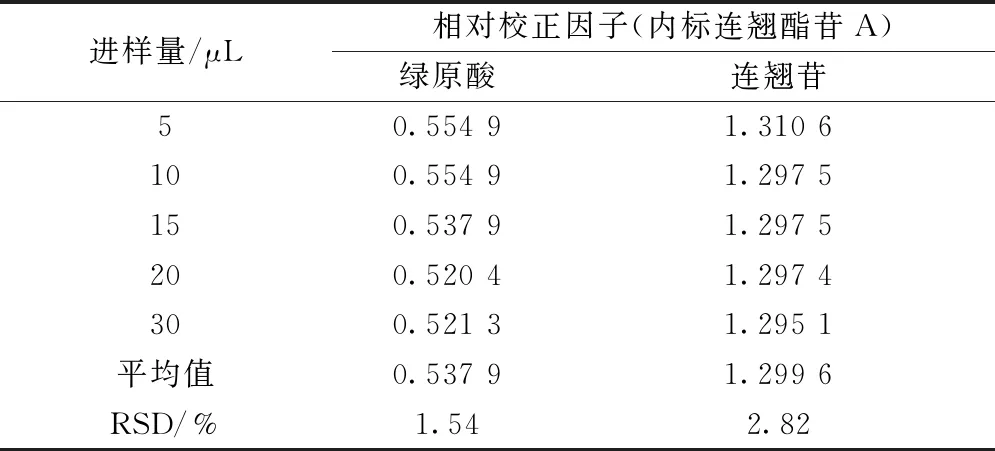
表2 各成分相对校正因子
2.10 不同仪器、色谱柱对相对校正因子的影响 分别采用Agilent 1260型、Waters e2695型色谱仪,以及Thermo Scientific BDS HYPERSIL C18、Hypersil BDS C18色谱柱(150 mm×4.6 mm,5 μm),按“2.9”项下方法计算相对校正因子,平行6次,结果见表3,可知均无明显影响(RSD<3%)。
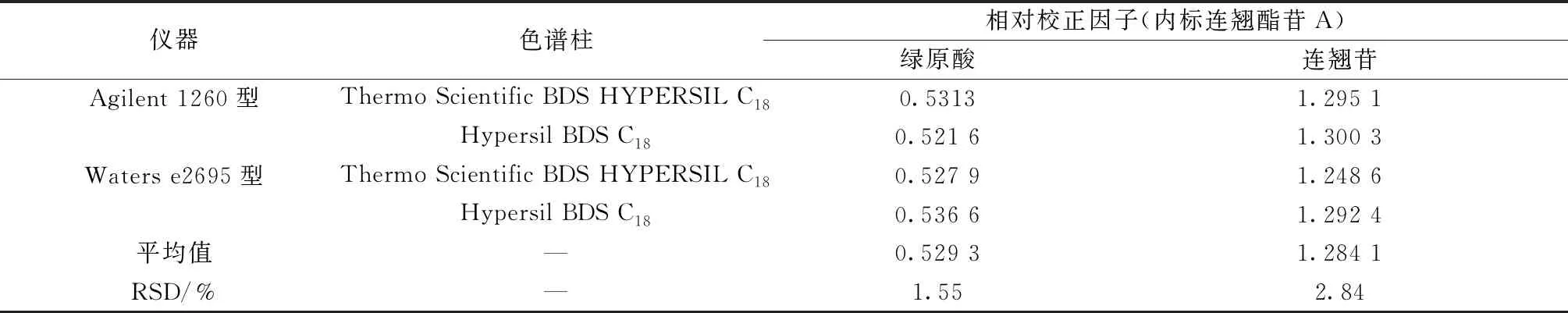
表3 不同仪器、色谱柱对相对校正因子的影响
2.11 色谱峰定位 考察了“2.10”项下仪器、色谱柱对相对保留时间的影响,结果见表4,可知均无明显影响(RSD<3%)。

表4 不同仪器、色谱柱对相对保留时间的影响
2.12 样品含量测定 取3批样品,按“2.2.1”项下方法制备供试品溶液,各平行3份,在“2.1”项色谱条件下进样测定,分别采用外标法、一测多评法计算含量,结果见表5。由此可知,2种方法所得结果接近[相对误差(RE)<1%],表明一测多评法可用于含量测定。
3 讨论
现代药理研究表明,连翘酯苷A、连翘苷具有抗菌、抗衰老等作用[10-12],绿原酸具有抗内毒素、抗肿瘤等作用[13-14],均与连翘归尾煎功能主治相符。由于连翘酯苷A在HPLC色谱图中的出峰位置位于绿原酸和连翘苷的中间,并且性质较稳定,故本实验以其为内标进行含量测定。
本实验考察了流动相乙腈-0.2%甲酸、乙腈-0.1%甲酸的分离效果,发现以乙腈-0.1%甲酸洗脱时各成分基线分离较好,符合实验要求。
4 结论
目前,有关一测多评法的报道大多是测定同一味药中同一种类成分的含量[15-16],而本实验测定了连翘归尾颗粒中不同种类成分的含量,并且该方法具有专属度高、重复性好、稳定性强、快速简便、节约成本等优点,可为该制剂质量控制和评价提供参考依据。
猜你喜欢
今日农业(2022年16期)2022-09-22
煤化工(2022年3期)2022-07-08
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
今日农业(2021年21期)2022-01-12
当代水产(2021年9期)2021-12-02
西北民族大学学报(自然科学版)(2020年4期)2020-12-21
今日农业(2020年19期)2020-12-14
今日农业(2020年16期)2020-09-25
广西植物(2020年5期)2020-07-04
山东工业技术(2016年10期)2016-09-06
