
De novo transcriptome assembly of Aureobasidium melanogenum CGMCC18996 to analyze the β-poly(L-malic acid) biosynthesis pathway under the CaCO3 addition
2023-01-03 11:31GenanWangHaisongYinTingbinZhaoDonglinYangShiruJiaChangshengQiao
食品科学与人类健康(英文) 2023年4期
Genan Wang, Haisong Yin,c, Tingbin Zhao, Donglin Yang, Shiru Jia,*, Changsheng Qiao,,*
a Key Laboratory of Industrial Fermentation Microbiology (Tianjin University of Science and Technology), Ministry of Education, Tianjin 300457,China
b Tianjin Engineering Research Center of Microbial Metabolism and Fermentation Process Control, College of Biotechnology,Tianjin University of Science and Technology, Tianjin 300457, China
c School of Bioengineering, Tianjin Modern Vocational Technology College, Tianjin 300350, China
d Tianjin Huizhi Biotrans Bioengineering Co., Ltd., Tianjin 300457, China
Keywords:De novo transcriptome analysis β-Poly(L-malic acid)Aureobasidium melanogenum
A B S T R A C T β-Poly(L-malic acid) (PMLA) is a water-soluble biopolymer used in food, medicine and other industries.To date, the biosynthesis pathway of PMLA has not been fully elucidated. In this study, we sequenced the transcriptom e of strain Aureobasidium melanogenum under 20 g/L CaCO3 addition. The resulting sequencing reads were assembled and annotated for the differentially expressed genes (DEGs) analysis and novel transcripts identification. The result indicated that with the CaCO3 addition, the tricarboxylic cycle(TCA) cycle and glyoxylate pathway were up-regulated, and it also found that a non-ribosomal peptide synthetase (NRPS) like protein was highly expressed. The DEGs analysis showed a high expression level of malate dehydrogenase (MDHC) and phosphoenolpyruvate carboxykinase (PCKA) in the CaCO3 group, which indicated a cytosolic malate activity. We speculated that the malate should be transported to or synthesized in the cytoplasm, which was then polymerized to PMLA by the NRPS-like protein, accompanied by the up-regulated TCA cycle providing ATP for the polymerization. Depending on the analysis, we assumed that an NRPS-like protein, the TCA cycle, and the cytosolic malate together are contributing to the PMLA biosynthesis.
1. Introduction
β-Poly(L-malic acid) (PMLA) is a highly soluble biopolymer consisting ofL-malic acid subunits ligated at the ester bond. Each unit contains a f lanking carboxyl group in itsα-position [1]. These highly accessible carboxyl groups enable PMLA to interact with positively charged molecules, resulting in an excellent capacity for payloads of various drugs and biologically functional groups [2]. In particular,after PMLA hydrolysis, it will release its malate subunits, which is an alternative method for malate production [3], enabling PMLA a huge potential application in the food industry.
Since the potential function of PMLA, many efforts have been paid to investigate its biosynthesis pathway [4-6], and the primary method is underlying the increasing production of its only precursor malic acid. This mechanism leads the PMLA-production-optimal research mainly focused on the relationship between PMLA and the tricarboxylic cycle (TCA), reductive tricarboxylic cycle (rTCA), and glyoxylate pathways. Meanwhile, researchers also conducted PMLA fermentation optimization experiments for the large-scale industrial application, including the screening of mutant strains, optimizing fermentation conditions, selecting suitable carbon sources, and adding growth factors [7-9].
Besides these pathways, some hypotheses suggested that there could exist a synthetase responsible for the ligation of malate subunits.It had been reported that newly synthesized PMLA contained labeledL-malate subunits that converted by the radioactive-labeledL-aspartate located in the cytoplasm, and this phenomenon indicated PMLA synthetase functioning in an extramitochondrial environment [10].The same study had also suggested that PMLA is synthesized from malyl-AMP by an enzyme complex consisting of a malyl-AMP ligase and transferase rather than the ligation of malyl-CoA [10].However, some researchers assumed thatL-malate must react with CoA-SH under the catalysis of thiokinase to formL-malyl-S-CoA, by which the PMLA is polymerized [3]. More recently, a novel PMLA synthetase gene has been proposed, which was reported responsible for encoding a non-ribosomal-peptide synthetase (NRPS) located in the plasma membrane with 6 transmembrane regions [11].
Since decades of research and effort, the understanding of the PMLA biosynthesis mechanism has advanced continually. To date,combined with the fast-developed sequencing technology, especially the next-generation sequencing (NGS), it could quickly dive into a deeper understanding of the biosynthesis mechanism. Moreover, with the help of NGS technology, RNA sequencing (RNA-Seq) is now the method of choice to study gene expression and identify novel RNA species, accompanied by low background noise and greater dynamic range for detection ability [12].
This study employed a yeast-like fungus,Aureobasidium melanogenum, which possesses high PMLA production ability under the CaCO3addition. Thede novotranscriptome assembled method was applied to measure differentially expressed genes (DEGs)and identify the novel transcripts that would carry out the PMLA biosynthesis process.
2. Materials and methods
2.1 Microorganism and medium
A. melanogenumCGMCC18996 was isolated in our laboratory and preserved in the China General Microbiological Culture Collection Center (Beijing, China, No. CGMCC18996). The strain was stored in potato dextrose agar (PDA) slants at 4 °C and subcultured every two weeks. The seed medium contained 60 g/L sucrose, 3 g/L yeast extract, 2 g/L succinic acid, 1 g/L (NH4)2SO4,0.4 g/L K2CO3, 0.1 g/L KH2PO4, 0.1 g/L MgSO4, 0.05 g/L ZnSO4,and 0.1% corn steep liquor (CSL,V/V). The fermentation medium contained 180 g/L sucrose, 35 g/L peptone, 0.1 g/L KH2PO4,0.3 g/L MgSO4, 0.5 g/L KCl, and 0.05 g/L MnSO4. Both seed and fermentation media were sterilized at 121 °C for 20 min before use.In this study, the CaCO3group contained an extra 20 g/L CaCO3in the primary fermentation medium compared to the control.
2.2 Fermentation conditions
The primary seed culture ofA. melanogenumCGMCC18996 was prepared by inoculating cells grown on solid medium into 500 mL Erlenmeyer flasks containing 300 mL seed culture medium and then cultured at 25 °C for approximately 40 h in a rotary shaker (IS-RDS3, Crystal Technology and Industries, Inc., USA).
Fed-batch fermentation parameters were investigated in a 5 L stirred tank fermenter (GRJB-5D, Zhenjiang Gree Co., Ltd., China)containing 3 L fermentation medium inoculated with 300 mL seed culture, and the fermentation medium was operated at 25 °C for 168 h with 500 r/min agitation speed and 1.3 VVM aeration rate.
2.3 Assay of PMLA production and fermentation parameters
Fermentation broth (10 mL) was collected at different time points and centrifuged at 15 000 r/min. The resulting supernatant (5 mL) was mixed with 5 mL 2 mol/L H2SO4and then neutralized at 110 °C for 11 h. After neutralization, the sample was analyzed by HPLC (L-2000,Hitachi Ltd., Japan) using a Prevail C18organic acid column at 25 °C eluted with 25 mmol/L KH2PO4at a rate of 1.0 mL/min. The PMLA concentration was determined by comparing the difference betweenL-malate concentrations before and after hydrolysis.
The residual sugar was measured by SBA-40E-Bioanalyzer(Shandong, China), and the cell density was determined via the method of dry cell weight (DCW). Before measurement, in the CaCO3group, the excess CaCO3in 10 mL fermentation broth was eliminated by HCl (3 mol/L). The fermentation broth (10 mL) was centrifuged at 5 000 r/min for 10 min, and the resulting precipitate was washed twice with phosphate buffer saline (PBS). After recentrifugation, the precipitates were dried overnight at 80 °C and then weighed.
2.4 Transcriptome sequencing
The transcriptome was sequenced by Novogene (Novogene Bio-Technology Co., Ltd., USA) with the Illumina NovaSeq 6000 platform, and the sequencing methods below are following their protocol.
2.4.1 RNA quantification and qualification
RNA degradation and contamination were monitored on 1%agarose gels, and the purity was checked by NanoPhotometer spectrophotometer (IMPLEN, CA, USA). The integrity of RNA was evaluated using the RNA Nano 6000 Assay kits of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Only the RNA samples that passed the threshold were selected for the next library preparation step.
2.4.2 Library preparation and sequencing
The sequencing libraries were generated with 1 μg RNA per sample using NEBNext UltraTM RNA Library Prep Kit for Illumina(NEB, USA) following the instruction guide, and the index codes were added to attribute sequences to each sample. Briefly, the protocol used poly-T oligo-attached magnetic beads to purify mRNA and applied fragmentation buffer (NEBNext Strand Synthesis Reaction Buffer) to fragment the purified mRNA. The first and second-strand cDNA synthesis, end repair, adenylation, and adaptor ligation were performed following Illumina’s instruction guide. The cDNA fragments of preferentially 250-300 bp length were purified with AMPure XP system (Beckman Coulter, Beverly, USA), followed by PCR amplification. Then, 3 μg USER Enzyme (NEB, USA) was used for size-selected, and the library quality was assessed on the Agilent Bioanalyzer 2100 system. After the cluster generation, the paired-end libraries were sequenced on Illumina NovaSeq 6000 with a paired 150 read length.
2.5 Quality control and data filtering
The raw reads (fastq format) in compressed (gzip format) files were obtained from the Novegene cloud release system (Novogene Co., Ltd., USA). All compressed files were verified by the MD5 string provided by the cloud system, and the raw reads were evaluated by FastQC-v0.11.9 [13]. All the single resulting fastqc files were converged into one file by multiqc [14] to evaluate each sample’s quality control results. Then each raw fastq file was filtered by tool Fastp-0.20.1 [15] with the default setting mode. After the fastp filtering, the resulting clean reads were evaluated by the same fastqc method illustrated above, and the samples passed the overrepresented sequence and the quality score (QC > 30) sections were set to the next step.
2.6 Transcriptome assembly and annotation
The transcriptome assembly was carried out by Trinity-v2.13.0 [16]following the Trinity’s protocol [17]. Besides all samples’ assembly,considering the repetitive sequencing reads and different gene expressions between each sample, the samples in the different groups were paired, which formed nine combinations (i.e., sample 1 in the CaCO3group paired with sample 1, sample 2, and sample 3 in the control group, respectively) to assemble the transcriptome. All assemblies were then evaluated by BUSCO-v4.1.4 [18] using the saccharomycetes lineage dataset.
The assembly’s 6 frames translation was conducted by transdecoder-v5.5.0, and the annotation was performed by Trinotate-v3.2.1 [19] following the instruction guide. The Pfam [20]and SUPERFAMILY [21] databases were used to conduct the protein function prediction, and the transmembrane domain prediction was conducted by TMHMM-2.0 [22].
2.7 Quantitative real-time PCR
Genes (9) that represented the different hypothesized PMLA biosynthesis pathway were selected to the quantitative real-time PCR(qPCR) measurement for verifying the transcriptome results, using glyceraldehyde 3 phosphate dehydrogenase (GAPDH) and glucose 6 phosphate dehydrogenase (G6PDH) as the internal reference genes. The corresponding primers were designed and presented in supplementary file (Table S1), and the gene expression levels were analyzed by StepOnePlusTMReal-Time PCR System (ThermoFisher,USA). The qPCR was performed with ChamQ SYBR qPCR Master Mix (Vazyme, China), and the cDNA was synthesized with HiScript III RT SuperMix for qPCR (Vazyme, China), following the instruction guide. The transcriptome and qPCR data comparison were performed by normalizing data by MinMax scalerwhich normalized data into [0,1].
2.8 Computing resources and data analysis
The computational analysis was performed by a cloud server with 32 CPUs and 192 RAM (Tencent Cloud, Shenzhen, China).
All the experiments in this study were conducted with three replicates. The command-line tools were executed in a Linux system(Ubuntu Server 16.04.1 LTS 64 bit), and the resulting data were analyzed by R studio (R version 3.6.3) and Jupyter Notebook.
3. Results
3.1 Effect of CaCO3 on PMLA production
The addition of 20 g/L CaCO3had a significant effect on PMLA production. As showed in Fig. 1a, the final PMLA production reached 42.91 g in the CaCO3group, which was about a 4-fold increase compared to the control (9.53 g/L). Fig. 1b showed both groups’biomass, which indicated that the amount of biomass in the CaCO3group was larger than that of the control after 72 h fermentation,and the biomass in the CaCO3group showed a rapid increase during 36 to 48 h. Meanwhile, Fig. 1c depicted both groups’ yield, presenting a larger yield observed in the CaCO3group, which indicated a 3 to 4-fold increase compared to the control. Fig. 1d showed the residual sugar consumption, and the result stated that residual sugar in the CaCO3group was consumed faster than that of the control group, and it was totally consumed at 144 h, 12 h earlier than the control.

Fig. 1 PMLA fermentation parameters were evaluated in a 5 L stirred tank fermenter. The CaCO3 group contained an extra 20 g/L CaCO3 in the primaryfermentation medium compared to the control group.
3.2 Transcriptome assembly and annotation
To evaluate the different sample combinations for transcriptome assembly, all samples’ assembly and paired sample assembly were employed in this study. Fig. 2 depicted the BUSCO evaluation result,which showed that all the assemblies obtained a high score. Among them, the CG1_CC3 assembly got the highest score and lowest missing (M), indicating that it could match more genes than others in the given saccharomycetes lineage dataset.
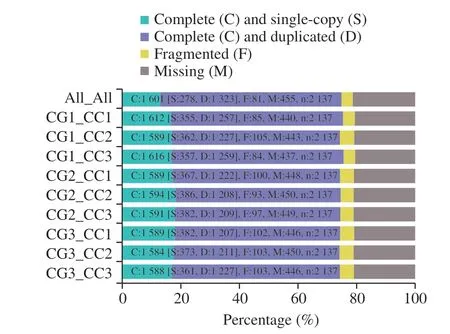
Fig. 2 Assembly evaluation by BUSCO, each combination was assessed by saccharomycetes lineage dataset. In the y axis, ‘All’ represents all samples; assembly, ‘CG’ represents the control group, and ‘CC’ represents the CaCO3 group. Each number after the ‘CG’ and ‘CC’ represents the replicate sample number. n: Total gene in the lineage.
The CG1_CC3 assembly was set to conduct the annotation process. After the database searching, the results found 19 980 genes and 39 335 transcripts in the annotation database, and the resulting CG1_CC3 assembly and its annotation were prepared for the following differentially expression and enrichment analysis.
3.3 DEGs and enrichment analysis
The DEGs analysis was conducted by tool DESeq2, and Fig. 3 showed the summarizing result of the DEGs of the CaCO3and control groups. The DEGs heatmap (Fig. 3a) summarized that 543 genes were differentially expressed in a large fold change (|log2FC| > 2).Among them, there were 248 up-regulated genes in the CaCO3group and 295 up-regulated genes in the control group. Figs. 3b and c showed the volcano plot and principal component analysis (PCA) plot with a small fold change (|log2FC| > 1). The result (Fig. 3b) indicated that 713 genes were up-regulated in the CaCO3group and 510 genes up-regulated in the control group. Meanwhile, the PCA plot (Fig. 3c)showed an apparent variation between both groups, in which thex-axis (PC 1) separated 73.5% of the features.
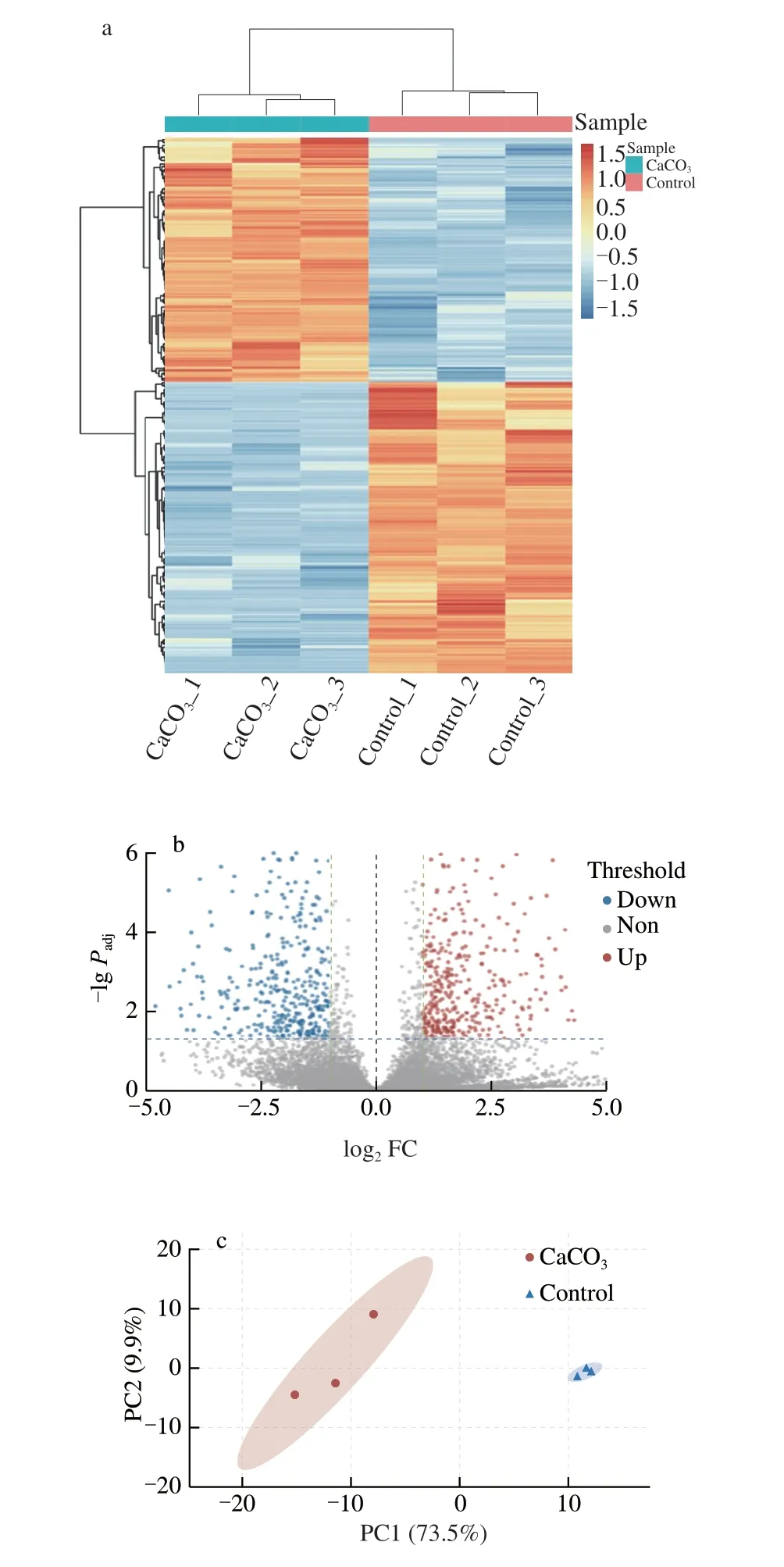
Fig. 3 (a) Heatmap of DEGs in a large fold change (|log2 FC| > 2), each row represents one gene, and the colors, orange to blue, represent the normalized value of TPM. (b) Volcano plot of DEGs in a |log2 FC| > 1 setting, color in red,grey, and blue represent up, non, and down regulated gene. (c) PCA plot of the samples, PC1 (73.5%) showed a clear variation of each group.
3.4 PMLA biosynthesis pathway
Three possible PMLA biosynthesis pathways and their related pathway were depicted in Fig. 4. The transcripts per million (TPM)value represented the genes’ relative concentration. Colored in red represented the up-regulated genes, and colored in black represented the genes that are not detected, down-regulated, or expressed at the same level. The results showed that, with the addition of CaCO3,most genes related to the TCA cycle,aco,idh1,idh3,dlsT,ogdH,lsc1,sdhA,fumH, andantwere up-regulated. The genes related to the glyoxylate pathway,aceAandaceB, were also expressed at a high level, which catalyzed a straight biosynthesis from isocitrate to glyoxylate and finally malate. Meanwhile, apparent changes were observed inastandmdhC, which encoded enzymes located in the intermembrane space of the mitochondrial or cytosol. These two genes are related to an aspartate-malate shuttle that functions as a malate transporter between the mitochondrial membrane. Moreover, an apparent TPM increase was observed in genepckA, which catalyzed a reversal reaction between phosphoenolpyruvate and oxaloacetate. The result showed thatpckAexpressed at around 50 000 TPM value, about a 4-fold increase compared to the control group, under the CaCO3addition.

Fig. 4 DEGs related to the PMLA biosynthesis pathway, up-regulated genes were colored in orange (gltA: Citrate synthetase, aco: Aconitate hydratase,idh1: Isocitrate dehydrogenase, idh3: Isocitrate dehydrogenase (NAD+),ogdH: 2-Oxoglutarate dehydrogenase E1 component,dlsT: 2-Oxoglutarate dehydrogenase E2 component, lsc1: Succinyl-CoA synthetase alpha subunit, sdhA: Succinate dehydrogenase,fumH: Fumarate hydratase, aceA: Isocitrate lyase, aceB: Malate synthase,pyc: Pyruvate carboxylase, mdh1: Malate dehydrogenase, mqo: Malate dehydrogenase(quinone), ast: Aspartate aminotransferase, mdhC: Malate dehydrogenase(cytoplasm), pckA: Phosphoenolpyruvate carboxykinase,ant: ADP/ATP antiporter. The orange and purple ovals represent malate shuttles).
Fig. 5 depicted the top 10 gene ontology (GO) classification with molecular function (MF) and biological process (BP) of the up-regulated genes in the CaCO3group. In order to select the determining DEGs that influenced the PMLA biosynthesis, the log2FC was set to 2 as the GO enrichment searching threshold. The results showed that the DEGs (log2FC > 2) were mainly involved in the pathways related to the transmembrane transporter activity, proton exchange, AMP binding, and phosphopantetheine binding activity. Moreover, after the repetitive gene filtering, the highly DEGs enrichment classification,such as binding activity and ligase activity, pointed out an enzyme complex highly expressed under the CaCO3addition.
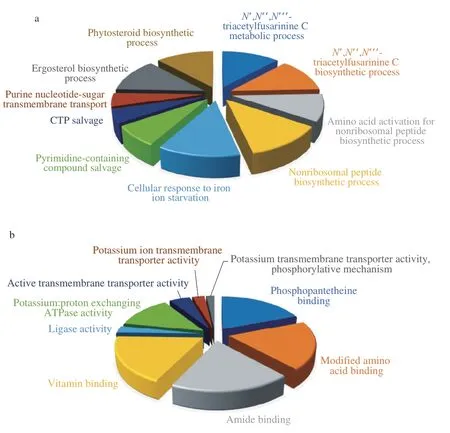
Fig. 5 GO enrichment was conducted by enriching DEGs in a large fold-change (|log2 FC| >2), and the top 10 enriched classifications were plotted.(a) BP classification, (b) MF classification.
To verify the transcriptome results, nine genes that represented the hypothesized PMLA biosynthesis were selected to conduct the qPCR experiment, in which gene,aco,ogdH,fumH, andantrepresented the TCA cycle,aceAandaceBrepresented the glyoxylate pathway, andmdhC,pckA, and the gene of the NRPS protein represented the cytosolic PMLA biosynthesis. The transcriptome and qPCR comparative results were shown in Table 1, and the normalized comparative plot was showed in Fig. 6, which indicated that two methods revealed the same relative trend in the comparison, but genes related to the glyoxylate pathway,aceAandaceB, expressed at a higher level with the qPCR methods.
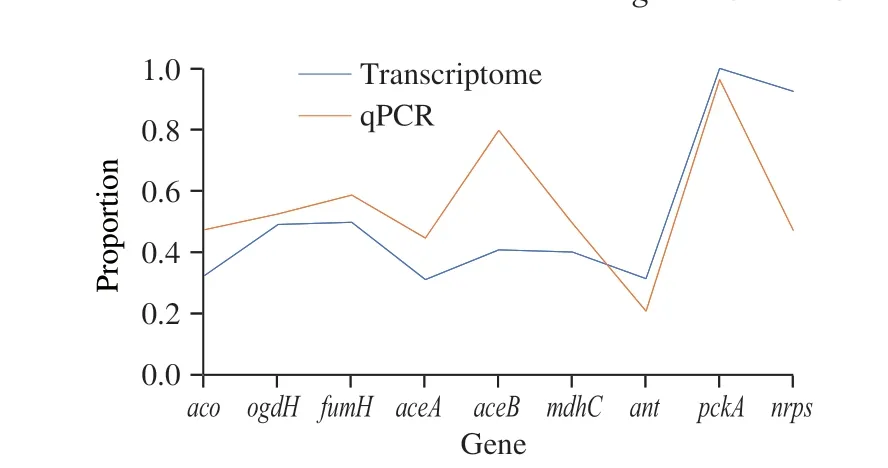
Fig. 6 Normalized data plot of qPCR and transcriptome results.

Table 1Comparison between qPCR and transcriptome results.
3.5 GO enrichment analysis
The GO analysis enriched a set of highly similar genes in phosphopantetheine, amide binding, modified amino acid binding,vitamin binding, and ligase activity (MF enrichment). The same result was also found in the BP enrichment. These highly similar gene sequences were collected as the search queries for BLASTx searching using non-redundant protein sequences (nr) database, and the results pointed to a peptide synthetase (Sequence ID: KEQ66003.1)in theA. melanogenumgenome (GenBank assembly accession:GCA_000721775.1). Directly searching (using BLASTn) these highly similar gene sequences in the same genome could not match a similar protein, but the gene location, which indicated the gene located in the genome scaffold 3 from position 1152275 to 1160499, 1160672 to 1167538, and 1152078 to 1152225. Gene length of these enriched genes and their similarity to the peptide synthetase (KEQ66003.1)were plotted in the Supplementary file (Fig. S1), and the results indicated that an assembly transcript, named Trinity_DN19_c0_g1(DN19), has 14 768 bp in length and shared an 88.4% similarity with the synthetase mentioned above (sequencing reads matching results were shown in the supplementary file (Fig. S2). Meanwhile, those genes with a highly similar sequence to the peptide synthetase were all sharing an identical sequence (100% in local alignment).

Fig. 7 Functional prediction of PMLA synthetase was conducted using the six-frame translation result of DN19 as the search query. The possible polymerization method was proposed, “A” represents the adenylation domain, “P(T)” represents the peptidyl carrier protein, and “C” represents the condensation domain. As the illustration shown, the condensation “C” domain might catalyze the reaction of -COOH and -OH, which formed an ester bond between malate acids. Upper right:the 3-dimensional crystal structure of DN19’s most BLASTP hit results, the crystal structure was retrieved from the Protein Data Bank (PDB entrance: 6MFZ).
3.6 Functional prediction of the peptide synthetase and the assembly DN19
The peptide synthetase and assembly DN19 mentioned above were prepared for the functional prediction using Pfam as the searching database. The results indicated a protein with about 5 000 amino acids that encoded several AMP binding enzymes, phosphopantetheine attachment sites (PP site), and condensation domains. Meanwhile, The TMHMM predicted that the protein does not have any transmembrane domain (Fig. S3), and it should locate in the cytoplasm based on its length. The PDB crystal structure with most BLASTp hit (Evalue =1.970 6 × 10-130, PDB entrance: 6MFZ) [23] was shown in Fig.7a, and the protein’s functional prediction results and its putative interaction with malate were depicted in Fig. 7b. Detailly, the malate should first be synthesized in or transported to the cytoplasm. Then, the AMP binding site (block A) caught the malate and catalyzed the formation of malyl-AMP from malate and ATP. The newly synthesized malyl-AMP was recognized by the PP site (block P), which ligated the malyl-AMP with its flanking sulfhydryl group, and then the malate was polymerized into PMLA in the condensation domain (block C),by which the malate conducted a dehydration condensation reaction.Further searching for the SUPERFAMILY database showed that all the domains (block A, P, and C) mentioned above assemble an NRPS-like protein that comprised of multiple A (block A), T (block P),and C (block C) domains function as a synthetase to recognize the cytosolic malate and catalyze the polymerization of PMLA.
4. Discussion
The researchers had already demonstrated that CaCO3strongly stimulated PMLA production [23]. Our previous study (data no show)also found that the addition of 20 g/L CaCO3had a considerable effect on PMLA production in strainA. melanogenum. As mentioned above,20 g/L CaCO3addition caused the PMLA production to increase about 4-fold, and an apparent increase in the yield was observed compared to the control group without CaCO3addition.
We sequenced the transcriptome of strainA. melanogenumwith and without CaCO3addition at the strain’s growing log phase (48 h)to figure out the mechanism, and the resulting sequencing raw reads generated by the NovaSeq 6000 platform obtained a high-quality score (Multiqc result was plotted in the supplementary file (Fig. S4).Thede novotranscriptome assembly method was then applied to assemble and annotate the transcriptome, and the resulting assembly was set to be used as the reference for the following DEGs analysis.In the assembly section, we employed the paired samples method,which means assembling one sample from the CaCO3group and one sample from the control group, and all samples’ assembly method,which means assembling all samples from both groups, to assemble the transcriptome for obtaining the best assembly that matches more gene in BUSCO evaluation.
For decades, many efforts have been paid to illustrate the PMLA biosynthesis pathway, and several pathways have been proposed [24],which were mainly related to the PMLA only precursor malic acid (i.e., the TCA cycle, glyoxylate pathway, and rTCA cycle).In this study, we investigate all three proposed pathways, and the results indicated that genes in the TCA cycle and glyoxylate pathway were up-regulated, especially for the TCA cycle, in which the genes had a higher TPM and log2FC value. We also found that geneant,encoding an ADP/ATP antiporter, was up-regulated, suggesting that more ATP was generated under the CaCO3addition. Some researchers [25] concluded that Ca2+could enter the mitochondrial matrix by a Ca2+uniporter, which could then activate the pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (IDH), and 2-oxoglutarate dehydrogenase (OGDH). In this study, all the genes responsible for these three dehydrogenases were up-regulated about 2 to 3-fold, indicating that the Ca2+signal might play a role in the up-regulated TCA cycle.
The results also showed that genes,astandmdhC, were highly expressed in the CaCO3group, suggesting that a malate/aspartate shuttle system might attribute to the malate transportation from the mitochondrial matrix to the interspace mitochondrial membrane. Some researchers found that Ca2+signal could activate aspartate-glutamate carriers (AGC) by its EF-hand Ca2+binding motifs [26].However, directly searching the malate/aspartate shuttle and EF-hand protein in the annotation dataset and DEGs do not show any apparent difference in the comparison, which indicated that malate generated by the TCA cycle may not account for the cytosolic malate accumulation. Further analysis showed that the expression level ofpckAin the CaCO3group increased considerably, and it encoded an enzyme, phosphoenolpyruvate carboxykinase (PCKA),that usually catalyzed the phosphoenolpyruvate formation from oxaloacetate [27]. However, researchers also indicated that PCKA is a bi-functional enzyme that could catalyze the reverse reaction from phosphoenolpyruvate to oxaloacetate [28]. Therefore, the highly expressedpckAmay contribute to the cytosolic oxaloacetate formation, which was then catalyzed to the cytosolic malate by malate dehydrogenase (MDHC). This mechanism directly synthesized malate in the cytoplasm and does not need the participation of the aspartate/malate shuttle system.
The glyoxylate pathway occurs in the glyoxysome, a mostly spherical organelle surrounded by a single lipid bilayer membrane.Some reported that filamentous fungi contain glyoxysome that is always accompanied by the characterized enzymes, malate synthetase and isocitrate lyase [29]. In the DEGs analysis, the genes responsible for these two enzymes were up-regulated about 2-fold under the CaCO3addition, strongly supporting the existence of glyoxysome inA. melanogenum. Our previous research (data no show) also showed that the addition of itaconic acid, an isocitrate lyase inhibitor, caused PMLA production to decrease by about 28%. To date, lack of the research focuses on the transport system of malate from glyoxysome to the cytoplasm, but some researchers suggested that glyoxysome may not have an aspartate/malate shuttle through proteomics analysis [30].Instead of the very-low permeability of the inner mitochondrial membrane, the single glyoxysome membrane might have a malate’s passive transport activity. This hypothesis could explain why the DEGs analysis could not match an apparent malate shuttle enzyme activity responsible for malate transportation. We then used qPCR to verify the transcriptome results. The comparison between the two methods indicated that these selected genes were presented in the same trend except geneaceAandaceB, which expressed higher with the qPCR methods. RNA-seq and qPCR followed two different protocols, and there could exist some dissimilarity. However, two methods all demonstrated that these genes were up-regulated in the CaCO3compared to the control.
In the GO enrichment analysis, we found an NRPS-like protein highly expressed in the CaCO3group. The NRPS protein is arranged in a modular structure that synthesized the secondary metabolites in microorganisms. It mostly consists of four domains,adenylation domain (A-domain) for activating the target substrate,peptidyl carrier protein domain (T-domain) for covalently lining of the activating substrate, condensation domain (C-domain) for the dipeptide formation, and a TE domain for cleavage of the full-length peptide product from the enzyme to terminate the biosynthesis [31].It has been reported that theA. melanogenumgenome contained 32 secondary metabolite biosynthetic clusters, and 2 of them are NRPS proteins [32]. In this study, the longest assembly transcript, DN19,possesses the characteristics of NRPS by functional prediction using Pfam. The result indicated several A, P(T), and C domains in DN19.Instead of forming peptide bonds like an NRPS, DN19 may catalyze the ester bond formation between the -COOH and -OH of each malate. Directly searching for the peptide synthetase (KEQ66003.1)using Pfam and SUPERFAMILY databases showed the same functional structure result as DN19 (in an A-T-C-A-T-C-T-C-A-T-CT-C-T-C arrangement), and this structure has also been reviewed as a hypothesized synthetase for PMLA polymerization [33]. Previous research has already demonstrated that an NRPS protein acting as a PMLA synthetase by knocking out its encoding gene, and it also stated that the Ca2+signal may play a major role [11]. However, in this study, it is the first time to detect the activity of this NRPS-like protein byde novotranscriptome analysis, and the TMHMM result showed that the NRPS-like protein (or DN19) in this study does not show any of the transmembrane domain, which is different from the previous study. We also presented the most BLASTp hit 3D-Crstal structure results of DN19 using biopython (version 1.7.8). However,we need more studies to dive into a deeper understanding of the role of the NRPS-like protein’s mechanism and its interaction with malate.
Briefly, malate should be first transported to the cytoplasm from mitochondrial matrix and glyoxysome, or it could be accumulated via PCKA-catalyzed cytosolic malate formation from phosphoenolpyruvate. Then, the cytosolic malate would be caught by the NRPS-like protein that catalyzed the formation of PMLA.The transcriptome analysis indicated that the mitochondrial malate transporters were expressed at a low level. Thus, the TCA cycle may not account for the cytosolic malate accumulation, but the glyoxylate pathway or the PCKA-catalyzed malate formation may contribute to this effect. In DEGs analysis, the up-regulatedantgene showed a high ADP/ATP exchange activity under the CaCO3addition,which could provide enough ATP for the NRPS-like protein to carry out the malate polymerization. As a result, we deduced that an NRPS-like protein that responsible for the PMLA polymerization, the up-regulated glyoxylate pathway and thepckAgene that responsible for the cytosolic malate accumulation, and the up-regulated TCA cycle that provided enough ATP for the whole process together contribute to the PMLA biosynthesis.
5. Conclusion
This study employed thede novotranscriptome assembly method to assemble theA. melanogenumtranscriptome for the following analysis. We proposed that the up-regulated glyoxylate pathway or genepckAmay account for the cytosolic malate accumulation,which was then polymerized by an NRPS-like protein. It is the first time to use thede novotranscriptome assembly method for strainA. melanogenum, and the results of this study presented a new insight for further study of the strain’s utilization and PMLA biosynthesis.
Conflict of interest
The authors declare there is no conflict of interest.
Availability of data and material
The sequencing data have been deposited in NCBI Sequence Read Archive (SRA) with the Bioproject accession PRJNA685601. The Transcriptome Shotgun Assembly (TSA) project has been deposited at DDBJ/EMBL/GenBank under the accession GIXY00000000. The version described in this paper is the first version, GIXY01000000.
Acknowledgements
The authors acknowledge the financial support of the Tianjin Municipal Science and Technology Commission (17PTGCCX00190,17PTSYJC00080, 17YFCZZC00310, and 16YFXTSF00460) and the Tianjin Engineering Research Center of Microbial Metabolism and Fermentation Process Control (ZXKF20180301).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://doi.org/10.1016/j.fshw.2022.10.007.

- 食品科学与人类健康(英文)的其它文章
- Emerging natural hemp seed proteins and their functions for nutraceutical applications
- A narrative review on inhibitory effects of edible mushrooms against malaria and tuberculosis-the world’s deadliest diseases
- Modulatory effects of Lactiplantibacillus plantarum on chronic metabolic diseases
- The role of f lavonoids in mitigating food originated heterocyclic aromatic amines that concerns human wellness
- The hypoglycemic potential of phenolics from functional foods and their mechanisms
- Insights on the molecular mechanism of neuroprotection exerted by edible bird’s nest and its bioactive constituents