
Neuroprotective effects of curculigoside against Alzheimer’s disease via regulation oxidative stress mediated mitochondrial dysfunction in L-Glu-exposed HT22 cells and APP/PS1 mice
2023-01-03 11:31WenqiWangYidiQuSiyuLiJinyuChuHongxinYangLirongTengDiWang
食品科学与人类健康(英文) 2023年4期
Wenqi Wang, Yidi Qu, Siyu Li, Jinyu Chu, Hongxin Yang, Lirong Teng, Di Wang*
School of Life Sciences, Jilin University, Changchun 130012, China
Keywords:Alzheimer’s disease Curculigoside Apoptosis Oxidative stress Mitochondrial dysfunction AMPK/Nrf2 signaling
A B S T R A C T Curculigoside (CCG) is a phenolic glycoside compound extracted from the root of a natural plant called Curculigo orchioides Gaertn. In this study, the neuroprotective effect of CCG through oxidative stress mediated mitochondrial dysfunction on L-glutamate (L-Glu)-damaged hippocampal neuron cell line(HT22) and APPswe/PSEN1dE9 transgenic (APP/PS1) mice were investigated. Observably, CCG in L-Glu-damaged HT22 cells suppressed apoptosis, reduced the accumulation of reactive oxygen species,balanced the mitochondrial membrane potential and prevented the over-inf lux of calcium. In APP/PS1 mice,4-week CCG administration significantly improved their memory and behavioral impairments, enhanced the function of cholinergic system, reduced the deposition of Aβ and neurofibrillary fiber tangles caused by tau phosphorylation, and suppressed the development and progression of oxidative stress in brains of APP/PS1 mice. Based on the screening of proteomic analysis on hippocampus, CCG were confirmed that it could regulate the expression levels of proteins related to mitochondrial dysfunction, mainly through activating on AMPK/Nrf2 signaling, in APP/PS1 mice and L-Glu-exposed HT22 cells. CCG has a prominent neuroprotective effect on regulate the AMPK/Nrf2-mediated mitochondrial dysfunction in cells APP/PS1 mice support CCG is a potentially potent drug for AD treatment and merits further investigation.
1. Introduction
Alzheimer’s disease (AD) is the “fifth killer” of the elderly population [1], characterized by memory loss and cognitive impairment. A survey suggests that 46.8 million AD patients have been diagnosed, with 4.6 million new cases per year, particularly in the Asia-Pacific region. AD is not only physically devastating to the sufferer, but also mentally and financially stressful to the family. The typical histopathological changes in AD are deposition of beta-amyloid (Aβ) in the brain and neurof ibrillary tangles (NFTs)dominated by phosphorylated tau protein [2]. Tau and Aβ work together to destroy the integrity of the nervous system, leading to the appearance of full-scale dementia in memory and consciousness [3].
Changes in mitochondrial structure and dynamics play a critical role in the pathological progression of AD. In the mitochondrial dysfunction response, mitochondrial dynamics imbalance and mitochondrial fragmentation increase, lead to mitochondrial DNA (mtDNA) damage, and then induce cell apoptosis [4]. The accumulation of Aβ and tau protein damages the mitochondrial integrity, reduces ATP production, and increases reactive oxygen species (ROS) content, which stimulates the production of Aβ and NFTs further [5]. Damaged mitochondria can release mtDNA and ROS,which enter the cytoplasm and trigger inflammatory responses [6].Oxidative damage occurs in the early stage of AD, prior to the deposition of Aβ [7]. ROS is produced through mitochondrial multi-step oxidative phosphorylation, which damages mtDNA and in turn accelerates mitochondrial oxidative phosphorylation. AMP-activated protein kinase (AMPK) is responsible for regulating intracellular metabolism and energy balance, and it’s highly sensitive to the AMP/ATP ratio [8], which can activate the phosphorylation of nuclear factor erythroid 2-related factor (Nrf2) [9]. When ROS attacks mitochondria, Nrf2 enters the nucleus and activates protective genes,to improve these genes expression level and prevent oxidative stress damage [10].
Cholinesterase inhibitors andN-methyl-D-aspartate (NMDA)receptor antagonists have been approved by the U.S. Food & Drug Administration [11,12]. However, due to the adverse effects on gastrointestine, tacrine (the cholinesterase inhibitor) was withdrawn from clinics in 2003 [13]. What’s more, memantine (NMDA receptor antagonists) will enhance the risk of fatigue, pain, confusion, urinary incontinence and urinary tract infection in the gerontal patient [14].Thus, people are turning their attention to plant-derived therapies for AD due to their extensive pharmacological activities with little adverse effects.Curculigo orchioidesGaertn, a natural plant belonging to the family Lycoris, distributed in the south of China, has various functions including immunoregulation, anti-inflammation,antibacterial and anti-tumor [15,16]. As the main active component ofCurculigo orchioidesGaertn, curculigoside (CCG) is a phenolic glycoside compound (Fig. S1). Current studies mainly focus on its antioxidant function in osteogenic injury and myocardial ischemia reperfusion [17,18]. It has been found that in primary cultured mouse cortical neurons, CCG protected the cells against the NMDA damage via regulating the levels of B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (Bax) [19]. CCG can facilitate fear extinction and prevent depression-like behaviors in mice related to regulation the brain-derived neurotrophic factor [20]. In APP/PS1 mice, CCG improved spatial learning abilities and memory and prevented bone loss mainly related to its anti-oxidative character [18].However, till now, few studies explicitly expose the neuroprotective properties of CCG against AD in both cells and animals, and the potential mechanisms.
Hippocampal neuron cell line, HT22, a sub-line separated from primary mouse hippocampal neuron cultures, is suitable for studying glutamate toxicityin vitro[21]. In this study, the neuroprotective effects of CCG onL-glutamate (L-Glu)-damaged HT22 cells and APP/PS1 mice were investigated. The relationship of the neuroprotective effect of CCG on AD and oxidative stress mediated mitochondrial dysfunction was analyzed through animal behavior,enzyme-linked immunosorbent assay (ELISA), proteomics, and western blot techniques. These results provide a theoretical basis for the protective effect of CCG on neurons in patients with AD.
2. Materials and methods
2.1 Cell culture and agent treatments
HT22 cells (No. BNCC337709, BeNa Culture Collection,Beijing, China), was grown in Dulbecco’s modified Eagle’s medium(DMEM) supplemented with 10% of fetal bovine serum (FBS), 1% of 100 µg/mL streptomycin and 100 units/mL penicillin at 37 °C in the existence of 5% CO2. All agents for cell culture were purchased from Gibco, Grand Island, NY, USA.
HT22 was incubated with 25 and 100 µmol/L of CCG (CAS:#85643-19-2; purity ≥ 98%; obtained from Shanghai Yuanye Bio-Technology Co., Ltd., Shanghai, China) for 3 h in serum-free medium, following by co-incubation of 25 mmol/L ofL-Glu (G8415,St. Louis, MO, USA) for another 24 h.
2.2 Flow cytometry assay
HT22 cells (3 × 105cells/mL) were seeded into 6-well plates.After 12-h incubation at 37 °C, cells were treated withL-Glu and CCG as described above, and then stained with Dead Cell Kit(MCH100105, Millipore, Billerica, MA) in darkness for 15 min at 37 °C. Cell fluorescence was detected by flow cytometry using a MuseTMCell Analyzer (Millipore, Billerica, MA).
2.3 Changes of mitochondrial membrane potential (MMP),ROS and Ca2+ detected by fluorescence probe
HT22 cells (1 × 105cells/mL) were seeded into 6-well plates.After 12-h incubation at 37 °C, cells were treated withL-Glu and CCG as described above. The analysis method for changes of MMP (JC-1,Millipore, Billerica, MA, USA), ROS (DCFH-DA; Nanjing Jiancheng Bioengineering Institute, Nanjing, China), and Ca2+(Fluo-4-AM,Molecular Probes, USA) was the same as what in our previous study [22].The immunofluorescence positive signals were analyzed and quantified by ImageJ Version 1.8.0 (National Institutes of Health,Bethesda, MD, USA). The CTRL group was used as the 100%benchmark, and other groups were compared with it.
2.4 Animal experimental protocol and drug treatment
The experiment was carried out with the approval of the Institution Animal Ethics Committee of Jilin University (SY201905015).Mice were purchased from Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China (SCXK (SU) 2015-0001)),and kept in a room with a constant temperature of (23 ± 2) °C.Other conditions include a humidity of 40%-60% with a 12-h/12-h light/dark cycle, and plenty of nutrients like water and food. Thirty B6C3-Tg (APPswePSEN1dE9)/Nju double transgenic male mice[Genotype: (Appswe) T, (Psen1) T] (APP/PS1) (8 months old, 45-50 g)were randomly divided into three groups, and given normal saline(20 mL/kg) serving as the vehicle group (n= 10), 10 mg/kg (n= 10)and 30 mg/kg (n= 10) of CCG through intragastric administration for 28 days. Another 10 wild-type male mice [Genotype: (Appswe) W,(Psen1) W] (WT) (8 months old, 45-50 g) were orally given normal saline (20 mL/kg) for 28 days serving as the control group. Finally,9 mice in each group were used for further detection. After the last behavioral test, the mice were euthanized by injecting 150 mg/kg barbiturate sodium (St. Louis, MO, USA). The serum together with tissues of the brain, spleen, and kidney were collected for biochemical and pathological analyses.
2.5 Behavior tests
2.5.1 Y maze test
Y maze has three arms of equal length, the angle between each arm is 120°, and the dimension of each arm is 47 cm × 16 cm × 46 cm(Any-mazeTM, Stoelting Co., Chicago, USA). The end of one arm provided food, allowing the mice to run from the start arm to the other two arms to find food. After three days of practice, the 12-15 h starved mice were put into the start arm to search for food in the other arm, and the movement track and searching time of mice were recorded with a video camera. The time it took to find the food was used as a measure.
2.5.2 Morris water maze test
The water maze test, in which mice were forced to swim and learn to find hidden platforms in the water, consisted of a Morris water maze (MT-200, Chengdu, China), a camera system, and an analysis system for the animal’s behavior trajectory (S7200, Chengdu Techman Software Co., Ltd., Chengdu, China). The water depth of the round pool is 40 cm, and there is a circular platform with a diameter of 9 cm in one corner, which is hidden under the water surface of 2 cm. Appropriate amount of milk powder is added to the water to make the pool opaque. Mice were trained for 5 min each day for 4 consecutive days, then placed in the same position facing the pool, and the time of escape latency in the incubation period of the hidden platform in 120 s was recorded.
2.5.3 Open field test
The open field experimental apparatuses consist of a sound-proof reaction box with a blackened inner wall and a camera directly above.The bottom of the box is 30 cm × 30 cm, and the central area is 15 cm × 15 cm. The mice were placed in the center of the bottom surface of the box, and its moving track and time spent in the center area were recorded by a camera. After each experiment, the dirt left by the previous mouse would be wiped clean to avoid the odor affecting the subsequent test.
2.5.4 Step down test
The platform experimental apparatus includes a platform box of 30 cm × 30 cm, an experiment box with power at the bottom, a platform in 5 cm diameter, and a platform video analysis software(Any-mazeTM, Stoelting Co., Chicago, IL, USA). A video device was installed above the experimental chamber to record the exploration track of the mice. During the 4-day training, the mice were put into an electrified jumping box to acclimate for 5 min, and then placed on the platform after 24 h after the end of the training period. The time of these mice’s first jumping from the platform within 3 min was recorded separately, and the movement routes of these mice in the experimental box were filmed.
2.6 ELISA
The collected hippocampus tissues were homogenized with normal saline, and the protein concentration was detected via a BCA kit (Millipore, Billerica, MA). The level of Aβ1-42(E20118),acetylcholinesterase (AchE) (E2148), acetylcholine (Ach) (E20535),CAT (E21414), acetyltransferase (ChAT) (E21422), SOD (E20348M),ROS (E20634), MDA (E20347M), GSH-Px (E20584) (Shanghai Yuanye Biological Technology Co., Ltd., Shanghai, China),Actl6B(KT0841M2),NTRK2(KT0848M2),PRKAa2(KT0858M2),PRKAG2(KT0849M2),EPB41L1(KT0856M2),SH2B1andKER1(Jiangsu Kete Science Co., Ltd., Jiangsu, China) in the serum and the hippocampus was analyzed by commercial kits according to the structures.
2.7 TUNEL assay and immunohistochemistry test
The same as our previous studies, TUNEL staining was used to analyze the apoptosis neurons, and immunohistochemistry test was used to analyze the changes of protein level in brains [23] in APP/PS1 mice including Aβ1-42(dilution of 1:800, 4 kDa, ab32136),tau (phospho S396 dilution of 1:4 000, 79 kDa, ab170892) and 4-hydroxynonenal (4-HNE, dilution of 1:200, 78 kDa, ab46545)(Abcam, Cambridge, MA, USA). Image J Version 1.8.0 was used for quantitative analysis.
2.8 Proteomics
Hippocampal tissue was cleaved and homogenized in RIPA (St. Louis, MO, USA) buffer containing a mixture of 2%phenylmethanesulfonyl fluoride (PMSF) (St. Louis, MO, USA) and 1% protease inhibitor (St. Louis, MO, USA). The samples were quantified through BCA protein assay kit (Waltham, MA, USA).Acetone was added to precipitate the protein, which was digested through trypsin, and the peptide sample was purified with sodium deoxycholate (SDC) (St. Louis, MO, USA). After desalination, the peptide was dispersed by nano-UPLC (EASY-nLC1200, Thermo Fisher Scientific, Waltham, MA, USA) and detected by Q-Exactive mass spectrometry (Thermo Finnigan, Silicon Valley, CA, USA).
The protein database is from the UNIPROT database (Uniprot_mouse_2016_09). The protein sequence and its inverted decoy sequence were used in MaxQuant (1.5.6.0) search. Statistical analysis was performed on the standardized quantitative results to obtain corresponding differentially expressed proteins. Each group of samples contained hippocampal tissues of 3 mice. Proteins with a protein expression ratio greater than 1.5 or less than 0.66 were defined as differential proteins. Subsequent Gene Ontology (GO), protein interaction analysis and display were performed [24].
2.9 Western blot
The HT22 cells were treated the same as section 2.1. The cell samples were prepared the same as hippocampal tissue. The hippocampal samples were prepared the same as section 2.8. The 40 µg proteins (4× loading buffer denaturation) were separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis,and then the proteins were transferred to polyvinylidene difluoride membrane (GE Healthcare Life Science, Beijing, China). After blocking with 5% BSA at 4 °C for 4 h, the membrane was then incubated with primary antibodies (Table S1) at 4 °C for 12 h and the secondary antibodies (Table S1) at 4 °C for 4 h. After TBST rinsing, an enhanced chemiluminescence (ECL) kit (Merck Millipore,Billerica, MA) and imaging system (Bio Spectrum 600, UVP company, Upland, CA, USA) were used to capture the bands, which were then analyzed and quantified by the ImageJ software.
2.10 Statistical analysis
Data were presented as the mean ± SEM. One-way analysis of variance (ANOVA) on top of post-hoc multiple comparisons (Holm-Sidak test) was used to analyze the differences and significance via DSS(version 25.0; IBM Corporation, Armonk, NY, USA).P< 0.05 was considered statistically significant in the difference.
3. Results
3.1 CCG protects the HT22 cells against L-Glu caused apoptosis
In HT22 cells incubated withL-Glu, CCG reduced the cell apoptosis (P< 0.01) (Fig. 1A), prevented the hyper intracellular ROS levels (P< 0.001) (Fig. 1B), enhanced the mitochondrial membrane potential (P< 0.05) (Fig. 1C) and suppressed the over-influx of Ca2+(P< 0.001) (Fig. 1D).

Fig. 1 CCG resisted the L-Glu caused apoptosis in HT22 cells. (A) CCG suppressed L-Glu-induced HT22 cell apoptosis. (B) CCG prevented the over-accumulation of ROS in L-Glu-exposed HT22 cells (magnification ×200, scale bar: 50 µm). (C) CCG ameliorated the situation of MMP decrease in L-Glu-exposed HT22 cells (magnification ×200, scale bar: 50 µm). (D) CCG suppressed the influx of Ca2+ in L-Glu-exposed HT22 cells (magnification ×200, scale bar: 50 µm). A1-A4. CTRL, L-Glu, L-Glu + 25 μmol/L CCG, L-Glu + 100 μmol/L CCG, respectively. Data were shown as the mean ± SEM (n = 6). ###P < 0.001 vs. CTRL cells, *P < 0.05, **P < 0.01 and ***P < 0.001 vs. L-Glu-treated cells.
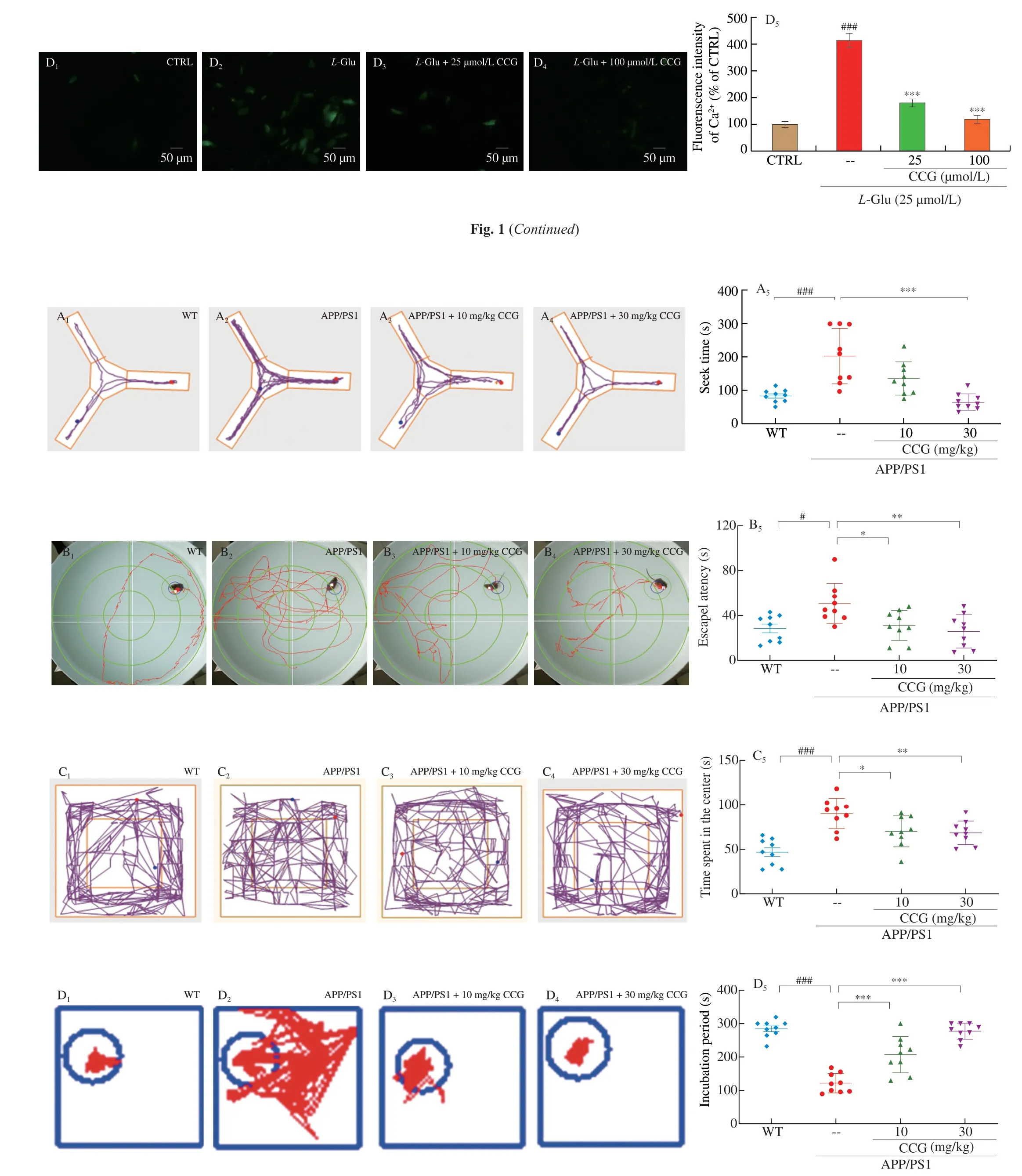
Fig. 2 CCG improved spatial recognition and memory in APP/PS1 mice. (A) CCG reduced the time taken to find food in the Y maze test of APP/PS1 mice (n = 9).(B) CCG shortened the time spent in seeking the platform of the Morris water maze test of APP/PS1 mice (n = 9). (C) CCG curtailed the time in the center of the open field test of APP/PS1 mice (n = 9). (D) CCG prolonged the time on the safe platform of step-down test of APP/PS1 mice (n = 9). CCG suppressed the level of (E) AchE (n = 6), and enhanced the level of (F) Ach (n = 6) and (G) ChAT (n = 6) in serum and hippocampus of APP/PS1 mice analyzing via ELISA. Data was shown as the mean ± SEM. #P < 0.05, ###P < 0.001 vs. WT mice, *P <0.05, **P < 0.01, ***P < 0.001 vs. APP/PS1 mice.
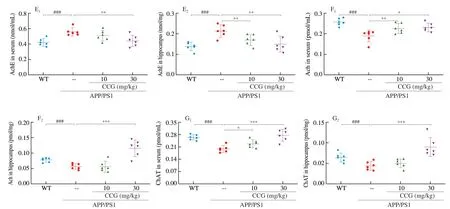
Fig. 2 (Continued)
3.2 CCG prevents cognitive decline in APP/PS1 mice
The autonomic exploration and learning behavior reflect the cognitive capacities of animals. The Morris water maze was carried out to evaluate the learning and memory ability of the mice in spatial position and direction. The Y maze can test the visual memory ability of mice. The open field test was performed to test the autonomous exploration behavior of mice in a new environment [22,23]. Step down test can evaluate the neurobehavioral phenomenon of mice after drug intervention [25]. The survival rate of all experimental mice was shown in Fig. S2. Compared with WT mice, the cognitive ability of APP/PS1 mice was impaired in the ethological tests (P< 0.05)(Figs. 2A-D). 28-day CCG administration shortened the time of getting food in the Y maze test (P< 0.001) (Fig. 2A), the time spent looking for a submerged platform in the Morris water maze test (P< 0.05)(Fig. 2B), the time spending in the central area of the open filed test(P< 0.05) (Fig. 2C), and prolonged the time on the safe platform in the step-down test (P< 0.001) (Fig. 2D) of APP/PS1 mice, respectively.Animal behavior experiments showed that CCG could significantly improve the learning and memory ability of APP/PS1 mice.
CCG administration showed that it has no significant effects on the bodyweights of APP/PS1 mice (Fig. S3).
The learning and memory capacity can be controlled by the central cholinergic system. Ach helps to maintain consciousness,and ChAT, a special marker of cholinergic neurons, are often used as an indicator to study cholinergic neurons [26]. Compared with non-treated APP/PS1 mice, CCG dose-dependently reduced the level of AchE (P< 0.01) (Fig. 2E), and enhanced the level of Ach (P< 0.05)(Fig. 2F) and ChAT (P< 0.05) (Fig. 2G) in serum and hippocampus.Accordingly, CCG may improve the learning and memory ability through the cholinergic system.

Fig. 3 (Continued)
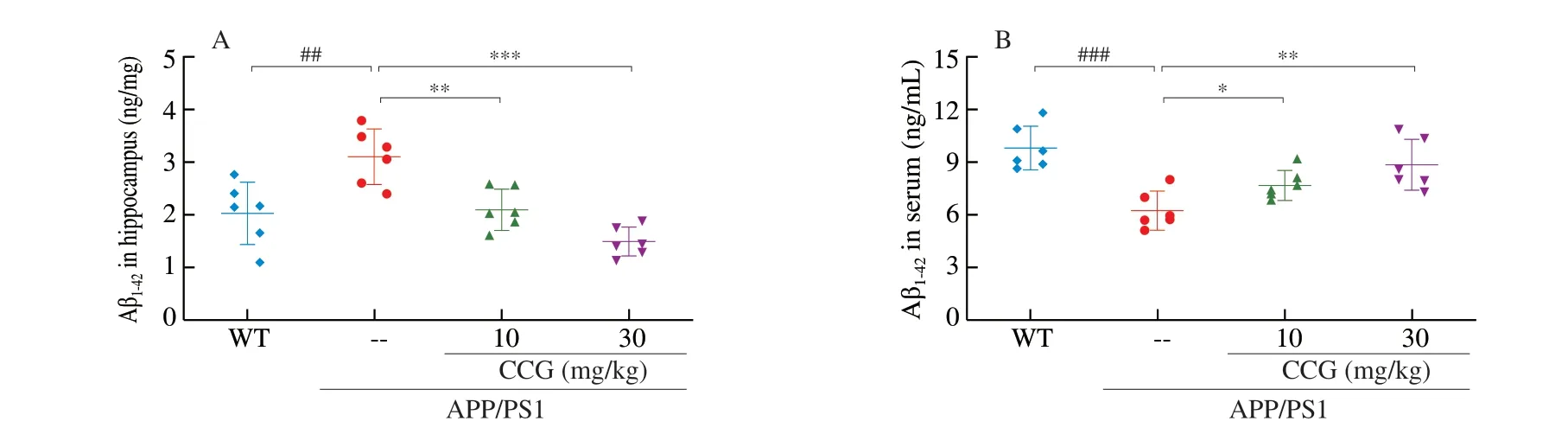
Fig. 3 CCG reduced the accumulation of Aβ and NFTs in brains of APP/PS1 mice. CCG (A) reduced the level of Aβ1-42 in hippocampus, and (B) enhanced the level of Aβ1-42 in the serum of APP/PS1 mice analyzing via ELISA (n = 6). (C) TUNEL staining showed that CCG significantly reduced apoptosis in the brains of APP/PS1 mice (magnification ×200, scale bar: 50 µm) (n = 4). (D) CCG reduced the expression of Aβ, and (E) the expression of p-tau in the brains of APP/PS1 mice detected by immunohistochemical staining (magnification × 100, scale bar: 100 µm) (n = 4). Data was shown as the mean ± SEM.## P < 0.01, ### P < 0.001 vs. WT mice, * P < 0.05, ** P < 0.01, *** P < 0.001 vs. APP/PS1 mice.
3.3 CCG prevents the deposition of Aβ and NFTs of APP/PS1 mice
Amyloid beta dimer acts as a toxic protein form to inhibit glutamate reuptake in neurons, leading to AD pathogenesis [27].During AD, abnormally phosphorylated tau protein loses its microtubule binding ability, resulting in neuronal tangles. In APP/PS1 mice, CCG significantly reduced the level of Aβ1-42in hippocampus lysis (P< 0.01) (Fig. 3A), and enhanced the level of Aβ1-42in serum(P< 0.05) (Fig. 3B) analyzed via ELISA. CCG significantly reduced the amount of TUNEL-positive apoptotic neurons in the brain of APP/PS1 mice suggesting the prevention on apoptosis after CCG administration(P< 0.001) (Fig. 3C). The amyloid plaque area of APP/PS1 mice was significantly larger than WT mice, while the pathological status was meliorated after CCG administration, suggesting Aβ aggregation was decreased by CCG (P< 0.001) (Fig. 3D).The excessive p-tau accumulation in APP/PS1 mice was strongly suppressed after 28-day CCG administration (P< 0.001) (Fig. 3E).
3.4 CCG reduces the oxidative damage in APP/PS1 mice
The free radicals in oxidative stress can promote the phosphorylation of Aβ and tau proteins [28]. 4-HNE, largely found in NFTs of AD patients, can promote abnormal phosphorylation of tau protein after modification [29]. In APP/PS1 mice, CCG significantly reduced the content of 4-HNE in brains (Fig. 4A). MDA is a specific marker of the peroxidation process among ROS and phospholipids and other macromolecules on biofilm. When the organism is subjected to oxidative stress caused by ROS, the organism itself has anti-ROS systems, such as GSH-Px, CAT, and SOD, to resist the peroxidation state [30]. Compared with non-treated APP/PS1 mice, CCG dose-dependently suppressed the level of ROS (P< 0.05) (Fig. 4B) and MDA (P< 0.05) (Fig. 4C), and enhanced the level of SOD (P< 0.01)(Fig. 4D), GSH-Px (P< 0.05) (Fig. 4E) and CAT (P< 0.01)(Fig. 4F) in serum and hippocampus lysis analysis via ELISA.CCG can suppress oxidative stress injury by reducing NFTs, ROS production, and increasing antioxidant content.

Fig. 4 CCG increased the anti-oxidative capacity in APP/PS1 mice. (A) CCG decreased the expression of 4-HNE in the hippocampus via immunohistochemical staining (magnification × 400, scale bar: 20 µm) (n = 4). CCG inhibited the production of (B) ROS and (C) MDA, and increased the activities of (D) SOD, (E)GSH-Px and (F) CAT in serum and hippocampus of APP/PS1 mice. Data was shown as the mean ± SEM (n = 6). #P < 0.05, ##P < 0.01, ###P < 0.001 vs. WT mice,*P <0.05, **P < 0.01, ***P < 0.001 vs. APP/PS1 mice.
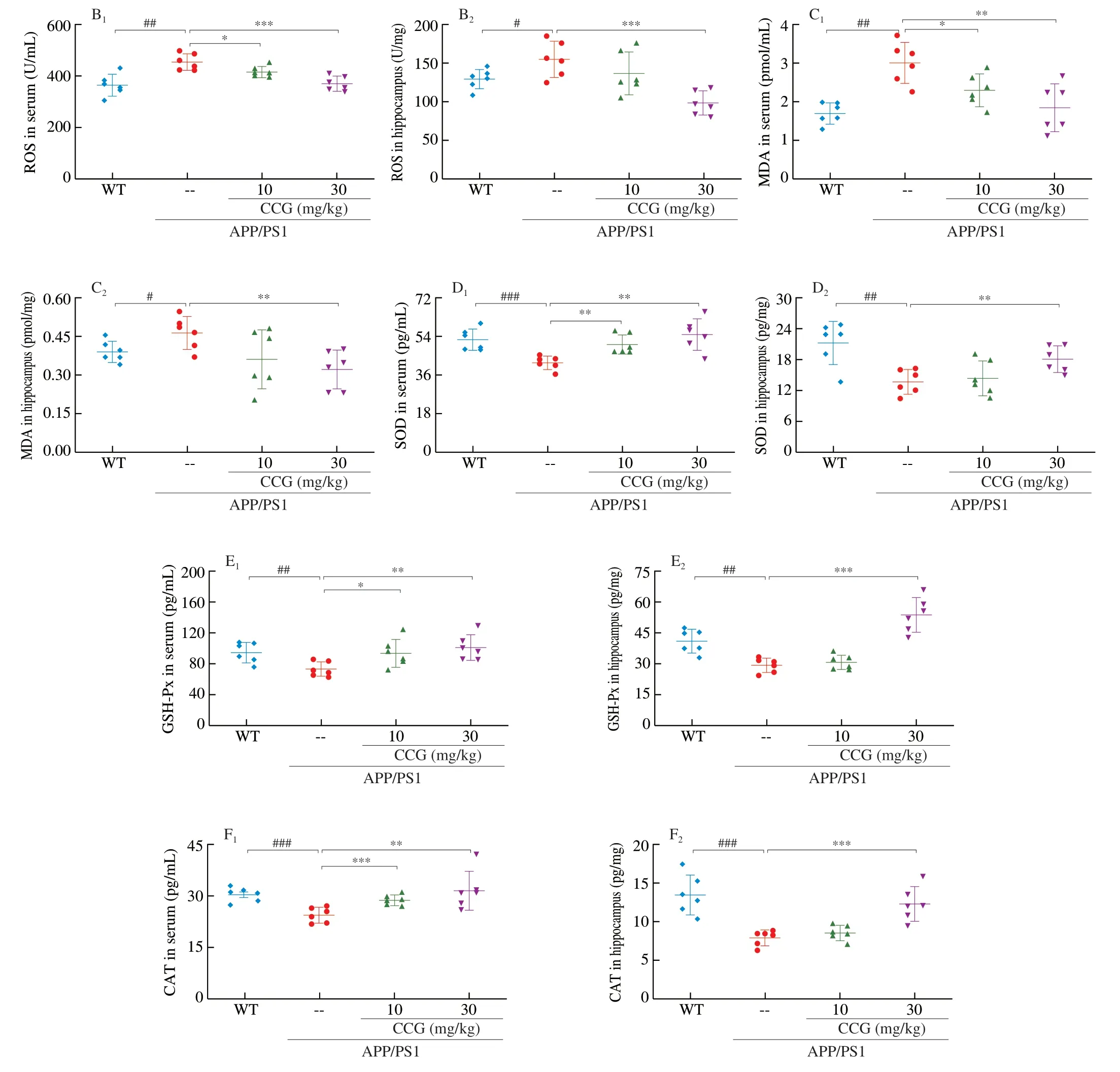
Fig. 4 (Continued)
3.5 CCG regulates the oxidative stress mediated mitochondrial dysfunction screened by proteomics
The unlabeled protein quantification technique is used for analyze the enzymatically hydrolyzed peptides of protein by mass spectrometry using liquid mass spectrometry technology.By comparing the signal strength of the corresponding peptides in different samples, the corresponding proteins can be relatively quantified [31]. Compared with non-treated APP/PS1 mice,28-day CCG administration suppressed 28 types of protein level and enhanced 39 types of protein level (Fig. 5A, Table S2). Meanwhile,among these factors, a total of 19 factors were associated with mitochondria, which have been highlighted in Table S2. The interactions among these changed proteins, mostly related to mitochondria, were represented with STRINGdb (Fig. 5B). According to GO enrichment analysis, for biological process, CCG regulated the cell morphogenesis, ribonucleoside metabolic process, neuron projection development, protein dephosphorylation and glycosyl compound metabolic process; for cellular components, CCG regulated excitatory synapse and mitochondrial inner membrane; for molecular function, CCG regulated protein kinase regulator activity which are all related to mitochondrial function (Fig. S4).
Furthermore, the significantly changed proteins in the hippocampus were confirmed via ELISA. Compared with non-treated APP/PS1 mice, CCG enhanced the level of Actl6B (P< 0.01)(Fig. 5C), NTRK2 (P< 0.01) (Fig. 5D), SH2B1 (P< 0.05) (Fig. 5E),EPB41L1 (P< 0.01) (Fig. 5F), KSR1 (P< 0.05) (Fig. 5G), and PRKAa2 (P< 0.01) (Fig. 5H), and reduced the levels of PRKAG2(P< 0.01) (Fig. 5I). These factors related to mitochondria could regulate the neural development and energy balance. Mutations in Actl6B cause synaptic loss [32], and synaptic mitochondrial dysfunction is often associated with microglia-related synaptic loss in AD [33].Cytoskeletal protein 4.1N expressed by EPB41L1 regulates the formation of Ca2+waves, an important mechanism that encodes Ca2+signals [34], which directly regulates mitochondrial dysfunction [35].The up-regulation of SH2B1 can reduce the inflammatory response,apoptosis and ROS level [36]. KSR1 is a key protein scaffold in ERK [37], helps to mediate mitochondrial morphological stability [38].NTRK2 regulates neurotrophic factors, resulting in defective axon transport, mitochondrial dysfunction and triggering mitochondrial dysfunction [39]. PRKAa2 and PRKAG2 also regulate AMPK, which

Fig. 5 CCG regulated proteins related to neural development and energy balance in hippocampus of APP/PS1 mice. The differences of protein expression in mouse hippocampus were analyzed by heatmap visualization and network analysis. (A) Differential protein heat maps among WT, APP/PS1 and CCG-treated APP/PS1 mice. The warmer the color, the higher the expression level (n = 3). (B) Protein STRING interaction diagram. Protein interaction networks were mapped to show the interactions among the significantly changed proteins. CCG regulated the level of (C) Actl6B, (D) NTRK2, (E) SH2B1, (F) EPB41L1, (G) KSR1, (H) PRKAa2 and(I) PRKAG2 in hippocampus lysis of APP/PS1 mice detected by ELISA (n = 6). Data was shown as the mean ± SEM. #P < 0.05, ##P < 0.01 vs. WT mice, *P <0.05,**P < 0.01, ***P < 0.001 vs. APP/PS1 mice.

Fig. 5 (Continued)

Fig. 6 (Continued)
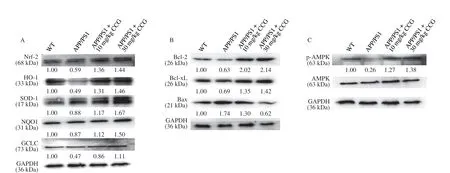
Fig. 6 CCG regulated the expression level of proteins related to mitochondrial stress in L-Glu-damaged HT22 cells and hippocampus of APP/PS1 mice. CCG enhanced the expression level of Nrf2 and its downstream proteins in (A) hippocampus of APP/PS1 mice and (D) L-Glu-damaged HT22 cells. CCG enhanced the expression level of Bcl-2 and Bcl-xL, and suppressed the expression level of Bax in (B) hippocampus of APP/PS1 mice and (E) L-Glu-induced HT22 cells.CCG enhanced the expression level of p-AMPK in (C) hippocampus of APP/PS1 mice and (F) L-Glu-damaged HT22 cells. Quantification data was normalized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and the corresponding total proteins, and were reported as the folds of those from the corresponding CTRL cells or WT mice (n = 4).
is involved in Aβ and tau phosphorylation in AD patients [40].
3.6 CCG regulates AMPK/Nrf2 signaling
At low level of oxidative stress, Nrf2-dependent antioxidant genes are activated, which can reduce ROS level to a level that is harmless to cells. InL-Glu-exposed HT22 cells and APP/PS1 mice, CCG enhanced the expression level of Nrf2 (P< 0.01) and its downstream antioxidative proteins including heme oxygenase-1 (HO-1) (P< 0.01),superoxide dismutase 1 (SOD-1) (P< 0.01), NAD(P)H quinone dehydrogenase 1 (NQO1) (P< 0.01), and glutamate-cysteine ligase catalytic (GCLC) (P< 0.01) (Figs. 6A, D, and S5A, B), suggesting the protection of CCG against the damage from oxidative stress [41].
In the process of mitochondrial dysfunction, along with mitochondrial apoptosis, Bcl-2 family proteins play a major role [42].InL-Glu-exposed HT22 cells and APP/PS1 mice, CCG enhanced the expression level of Bcl-2 (P< 0.01) and B-cell lymphoma-extra-large(Bcl-xL) (P< 0.01), and reduced the expression level of Bax (P< 0.01)(Figs. 6B, E, and S5A, B).
Phosphorylated AMPK reduces the production of anti-inflammatory factors [43] and promotes nuclear translocation of Nrf2 [9]. InL-Glu-exposed HT22 cells and APP/PS1 mice, CCG enhanced the expression level of p-AMPK (P< 0.001) (Figs. 6C,F, and S5A, B).
4. Discussion
Mitochondrial dysfunction at the early stage of AD can cause Aβ production and aggregation [44]. However, with the development of AD and the increase of Aβ and NTFs, the mitochondrial fragments and abnormal dynamics produce excessive ROS [45]. CCG therapy promoted the pathological progress of AD further, restored mitochondrial membrane potential drop, and calcium overload in cells, revived the disposition of Aβ1-42and phosphorylated tau tangles in brains of APP/PS1 mice, and regulated the expression level of Nrf2 and its downstream proteins, suggesting the important roles of oxidative stress mediated mitochondrial dysfunction in the CCG-alleviated AD symptoms.
The dimer of amyloid β is the most important form of a toxic protein, which can inhibit glutamate reuptake and impair glutamate homeostasis [27]. Consequently, Ca2+channels remain opening,and large amounts of Ca2+flow into the cell, prompting the cell membrane to enter autolytic state. Excessive Ca2+also passes through the mitochondrial membrane, destroys the normal mitochondrial electron transport chain and interfere with the process of oxygen consumption. Instead, a large number of by-products of ROS attack the mitochondrial membrane [46]. The permeability of mitochondrial inner and outer membrane is double regulated by Bcl-2 family proteins that resist and promote apoptosis [47]. When the apoptotic factor Bax is expressed in mitochondria, anti-apoptotic proteins such as Bcl-2 and Bcl-xL from the outer membrane will promote the formation of heterodimer of Bax to reduce its apoptotic activity [48].Bcl-2 itself is not an antioxidant, but acts as an oxidant to promote oxidative stress in cells and induce the enhancement of endogenous antioxidant capacity of cells, such as increasing activities of SOD,GSH-Px, CAT and other enzymes, to prevent mitochondrial dysfunction [49]. InL-Glu-damaged HT22 cells, CCG has positive effects on regulating oxidative stress mediated mitochondrial dysfunction and apoptosis.
In APP/PS1 double transgenic mice, the mutation of APP resulted in a change in its binding site to theβ-secretase, and the mutatedPS1gene changes the activity ofγ-secretase and alters the metabolic process of APP, causing the production and increase of Aβ1-42, and strongly simulating the formation of neuritic plaques in the brain of patients with AD [50]. Aβ deposition may lead to morphological and dynamical changes in mitochondria. Mitochondria produce stress response in order to maintain their shapes, structures and functions.The excess of ROS is responsible for the misfolding and aggregation of internal proteins. Ach factor protects cells from damage by inhibiting mitochondrial ROS production and reducing mitochondrial unfolded protein response [51]. On the basis of improving the cognitive ability of APP/PS1 mice, CCG significantly increased the contents of Ach and ChAT, and decreased the content of AChE,confirming the central role of Ach during the modulation of CCG on mitochondrial dysfunction. Furthermore, microtubule disintegration and synaptic degeneration caused by tau hyperphosphorylation affect the synthesis of neurotransmitters [52], especially choline ChAT and Ach, thus inducing the accumulation of AChE.
The process of mitochondrial dysfunction is related to oxidative stress, which leads to oxidative phosphorylation damage in mitochondria, and ultimately leads to neuronal damage and cognitive decline, a series of early clinical manifestations of AD [53,54].As a specific marker of oxidative reaction in AD patients, 4-HNE accumulates in the early stage of AD, disrupts mitochondrial balance and causes oxidative stress [55]. 4-HNE can accelerate the deposition of Aβ [56], inhibit the formation of microtubules, and promote the phosphorylation of tau protein which leads to the accumulation of NFTs. In APP/PS1 mice, CCG not only reduced the expression of 4-HNE in the hippocampus, but also regulated the enzymes relating to the oxidative stress. Under pathological conditions, the equilibrium state of redox is destroyed, resulting in the decrease of SOD, GSH-Px,and CAT [57], and the rapid accumulation of ROS and the oxidation end product MDA [58]. SOD is the most important antioxidant in mitochondria. The decrease of SOD content increases APP and produces toxic Aβ [59]. The decrease of antioxidant enzyme content in mitochondria and the surge of active oxides are bound to cause oxidative stress. CCG inhibits the generation of peroxides, regulates oxidative stress, and then regulates mitochondrial dysfunction.
As an important regulator of redox homeostasis in cells, Nrf2 regulates a series of antioxidant proteins and keeps cells in a stable state. Nrf2 controls NQO1, which can protect DNA from oxidative damage [41], HO-1, which can effectively resist ROS activity [60],GCLC, which affects GSH synthesis and protects against oxidative stress damage [61], and SOD. At the 550 serine residue of Nrf2 protein is directly catalyzed by activated AMPK [9] to regulate the expression level of detoxifying enzyme genes, thus activating the cell protection and improving the ability of anti-oxidative stress.AMPK interacts with Nrf2 and other factors to cause mitochondrial DNA replication and transcription, which affects mitochondrial synthesis [62]. When AMPK is deficient, metabolic balance will be disrupted and damaged mitochondria will accumulate,leading to mitochondrial dysfunction. AMPK can induce strong tau phosphorylation, and the Ser262may be the main phosphorylation site of AMPK on tau [63]. The genesPRKAG2andPRKAa2are related to the AMPK protein pathway and regulate cellular energy metabolism [64].PRKAG2gene expresses γ2 non-catalytic subunits on AMPK and mediates binding to AMP, ADP and ATP; meanwhile,PRKAa2is a gene expressing the catalytic subunit α2 of AMPK. The expression ofPRKAG2gene is positively correlated with the level of Aβ [40]. In response to mitochondrial dysfunction, CCG can save neuronal loss caused by AD by reducing Aβ deposition and NTFs, reducing the production of oxides, increasing the content of antioxidant enzymes,activating the AMPK/Nrf2 pathway to resist oxidative stress damage caused by mitochondrial dysfunction.
There are still limitations in present study. Based on the proteomics screening, other signals are involved in CCG-mediated anti-AD effects, which need further investigation. Accordingly,inflammation is responsible for the production of Aβ during the development of AD. CCG has been proved to possess the anti-inflammatory activity [65]. The effects of CCG on the neuro-inflammation will be analyzed in our further experiments.
5. Conclusions
This study is the first to demonstrate the neuroprotective effect of CCG onL-Glu-injured HT22 cells and APP/PS1 mice. CCG responds to oxidative stress mediated mitochondrial dysfunction by enhancing the expression level of antioxidant proteins, and alleviates mitochondrial dysfunction by activating the AMPK/Nrf2 signal to reduce the expression level of pro-apoptotic proteins in Bcl-2 family members (Fig. S6). Our data provide experimental evidence to investigate the clinical application of CCG as a potential agent to inhibit or reverse the progression of AD further.
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
This work was supported by the Science and Technology Develop Project in Jilin Province of China (20200201030JC), the Scientific Research Project of Education Department of Jilin Province in China (JJKH20211461KJ) and Characteristic Innovation Project for Guangdong University of China (2019KTSCX221).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The experiments complied with the ARRIVE guidelines and were carried out in accordance with National Institutes of Health guide for the care and use of Laboratory animals, and obtained an ethical review form regarding animal welfare approved by the Experimental Animal Center of Jilin University (License No. SY201905015).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://doi.org/10.1016/j.fshw.2022.10.009.

- 食品科学与人类健康(英文)的其它文章
- Emerging natural hemp seed proteins and their functions for nutraceutical applications
- A narrative review on inhibitory effects of edible mushrooms against malaria and tuberculosis-the world’s deadliest diseases
- Modulatory effects of Lactiplantibacillus plantarum on chronic metabolic diseases
- The role of f lavonoids in mitigating food originated heterocyclic aromatic amines that concerns human wellness
- The hypoglycemic potential of phenolics from functional foods and their mechanisms
- Insights on the molecular mechanism of neuroprotection exerted by edible bird’s nest and its bioactive constituents