
亚甲基四氢叶酸还原酶缺乏症致脑积水患儿2 例临床及MTHFR 基因变异分析
2023-02-27 04:28陈哲晖宋金青李梦秋张宏武姚红新杨艳玲
临床儿科杂志 2023年2期
董 慧 陈哲晖 马 雪 张 尧 宋金青 金 颖 李梦秋 张宏武 姚红新 杨艳玲
北京大学第一医院1.儿科;2.小儿外科(北京 100034)
高同型半胱氨酸血症是一组常见的代谢异常,病因复杂,已知多种遗传和非遗传病因,可自胎儿到老年发病,人群总体患病率高达5%,是可防可治的疾病[1-2]。遗传性高同型半胱氨酸血症1型被列入我国第一批罕见病[3],需要通过低蛋氨酸饮食、维生素B6、甜菜碱或肝移植治疗[4-5]。亚甲基四氢叶酸还原酶(methylenetetrahydrofolate reductase,MTHFR)缺乏症(OMIM 236250)所致同型半胱氨酸血症2型是较常见的类型,个体差异显著,多在青少年或成年发病,少数严重者在婴儿期发病,导致早发脑病[1],致残,甚至致死。
脑积水是一种严重的结构性脑损伤,早在新生儿期到婴儿期即可发病,甚至于胎儿期可能通过超声、磁共振扫描等检出,若不早期治疗,将引起不可逆性脑损伤,导致智力运动障碍及视力残疾,严重时致死[5-6]。一些同型半胱氨酸血症患儿合并脑积水,需多学科联合防控,在代谢干预治疗的同时进行侧脑室引流等手术治疗,以改善预后[7-8]。目前,国内外较少有关同型半胱氨酸血症2型引起脑积水的报告[9]。现回顾性分析2 例就诊于北京大学第一医院儿科以婴儿期脑积水为突出表现的患儿,并复习相关文献。
1 临床资料
例1,男,8月龄时因重度发育落后、间断抽搐1月余于2020年1月就诊。患儿自2月龄起纳差、反应迟钝,外院头颅MRI检查发现脑积水(图1A),进行侧脑室-腹腔分流术。7 月龄起出现癫痫发作,每日发作数次。口服托吡酯、左乙拉西坦及氨己烯酸等多种抗癫痫发作药物治疗,癫痫未能控制。
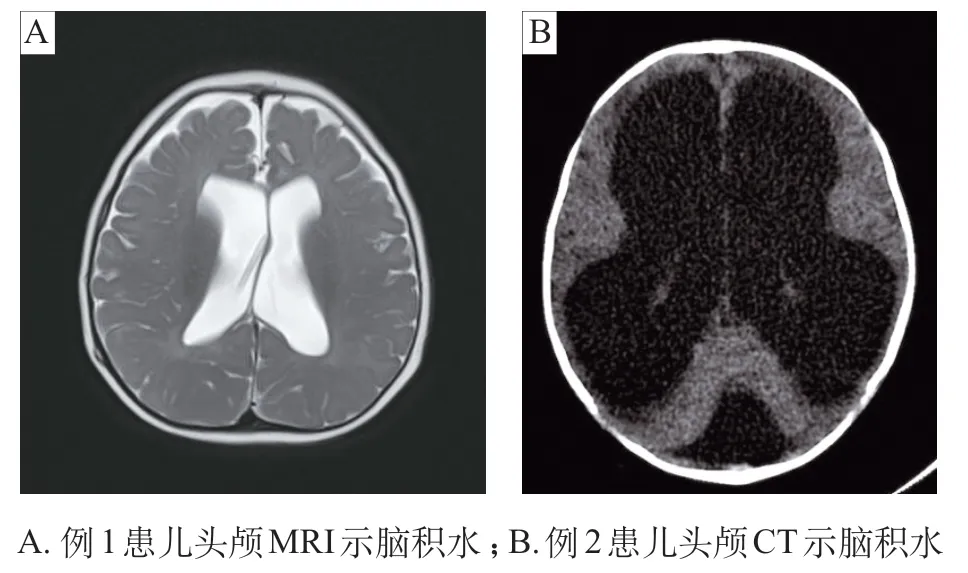
图1 患儿头颅影像学表现
患儿系G1P1,足月顺产,出生体重3 650 g,混合喂养。6月龄抬头,7月龄可稍坐及无意识发“baba、mama”音。患儿父母健康,非近亲婚配。体格检查:追声追物差,头围46.0 cm,前囟1 cm×1 cm,张力不高,右侧顶枕区及下腹部可见手术瘢痕,四肢肌力肌张力正常,可短暂扶站,深浅反射对称引出,病理征阴性。实验室检查:血常规、血氨、肝肾功能正常。血清总同型半胱氨酸显著增高,波动于65~102.6 μmol/L(正常参考值<15 μmol/L)。血液甲硫氨酸降低或处于正常低值(7.81~11.5 μmol/L,正常值10~50 μmol/L),甲硫氨酸/苯丙氨酸降低或处于正常低值(0.16~0.61,正常值0.2~1.4)。尿液甲基丙二酸等有机酸正常。心电图、超声心动图、腹部B超未见异常。视频脑电图可见不典型高度失律,左右半球有时不同步,监测到频繁孤立及成串痉挛发作。
例2,男,4月龄时因阵发性青紫14天于2017年12月就诊。患儿哭闹后青紫,面色发绀,伴抽搐发作,口唇及下颌抖动,双眼凝视,持续2~3分钟后缓解,止惊治疗后好转,约2~3 天发作1 次,外院头颅CT及MRI发现严重脑积水(图1B)。
患儿系G3P3,足月剖宫产,出生体重3 200 g。患儿2 个同胞姐姐均于2 月龄时夭折,大姐脑发育不良,二姐患肺炎合并脑积水。患儿为母乳喂养,智力运动发育迟缓,4 月龄时仍不能竖头、翻身,不能追声、追视,不易逗笑。体格检查:头围44cm,前囟饱满,直径约2×3 cm,双侧膝腱反射亢进,双侧巴氏征阳性,双侧踝阵挛阳性。实验室检查:血常规、肝肾功能、血氨正常。血清总同型半胱氨酸显著增高(>100 μmol/L),血液甲硫氨酸降低或处于正常低值(9.84~12 μmol/L),甲硫氨酸/苯丙氨酸降低或处于正常低值(0.18~0.28)。尿液甲基丙二酸浓度正常。心电图、胸片及腹部B超大致正常,超声心动图提示卵圆孔未闭。视频脑电图可见高度失律,双侧半球放电分离,后头部为主快波节律阵发,左侧为著。
结合2 例患儿临床表现及实验室检查,考虑为同型半胱氨酸血症2 型,合并脑积水、癫痫、全面发育迟缓。
监护人签署知情同意书后,取患儿及其父母静脉血各2 mL,检测患儿全外显子组序列。结果2 例患儿的MTHFR基因均检出复合杂合变异,采用一代测序方法检测患儿父母MTHFR基因,分别检出1个杂合变异。2例患儿确诊为MTHFR缺乏症所致同型半胱氨酸血症2型。
例1的MTHFR基因检出c.1915A>C(p.T639P)和c.568 T>C(p.F 190 L),均为未报道的新变异,c.1915A>C来源于父亲,导致MTHFR多肽链第639位氨基酸由苏氨酸改变为脯氨酸;c.568T>C来源于母亲,导致该多肽链第190 位由苯丙氨酸改变为亮氨酸。例2 的MTHFR基因检出c.1530 G >A(p.K510=)和c.233C>A(p.S78Ter),c.1530G>A来自母亲,为已报道的致病性变异[10],c.233C>A为未报道的新变异,来源于父亲,导致该多肽链第78位发生提前终止(图2)。
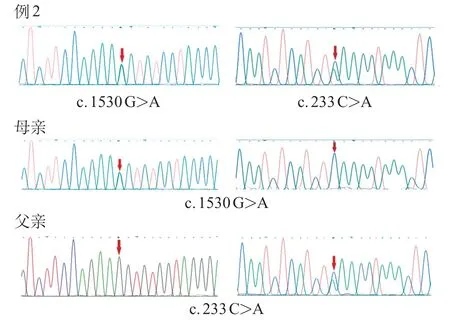
图2 例2 及其父母MTHFR 基因部分测序图
根据美国医学遗传学与基因组学学会发布的变异解读指南,分析3 个新变异的致病性[11],在人类基因变异数据库(HGMD、Clinvar)及千人基因组计划数据库(www.1000genomes.org)中未检出两患儿MTHFR基因3个新变异c.1915A>C、c.568T>C和c.233C>A。经Mutation Taster及UCSC Genome Browser 网站致病性预测及多物种同源性分析,c.1915 A>C、c.568 T>C 在哺乳动物中高度保守,c.233 C >A 引起氨基酸翻译的提前终止,均可能致病。
2 例患儿均转儿外科行侧脑室-腹腔分流术评估、治疗及随访。同时进行代谢干预及对症治疗。
例1 肌注维生素B121000 μg/w,口服甜菜碱1.5 g/d,亚叶酸钙 7.5 mg/d,维生素B660 mg/d,左卡尼汀 500 mg/d,代谢状态有所改善;继续抗癫痫治疗,口服托吡酯、左乙拉西坦及氨己烯酸,并生酮饮食。目前患儿3 岁,血清总同型半胱氨酸波动于70.6~85.8 μmol/L,血甲硫氨酸波动于11~15 μmol/L,仍有较频繁抽搐发作,智力运动发育迟缓。
例2 肌注维生素B12500 μg/w,口服甜菜碱1 g/d,亚叶酸钙 7.5 mg/d,维生素B640 mg/d,左卡尼汀 500 mg/d,代谢状态改善;同时应用左乙拉西坦。患儿1岁后失访。
2 讨论
高同型半胱氨酸血症病因及临床表现复杂,需要个体化治疗。根据血中总同型半胱氨酸浓度,高同型半胱氨酸血症分为轻型(15~30 μmol/L)、中间型(31~100 μmol/L)和重型(>100 μmol/L)[3]。采用液相串联质谱法行新生儿筛查,可检出同型半胱氨酸血症1型及甲基丙二酸血症合并同型半胱氨酸血症等部分患者[4,6],但是同型半胱氨酸血症2 型需通过生化代谢检查、基因检测等综合分析才能诊断[13],如果能早期诊治,绝大部分患者预后良好[3]。
MTHFR 是甲硫氨酸代谢过程中的关键酶,MTHFR缺乏症导致5,10-亚甲基四氢叶酸还原为5-甲基四氢叶酸过程受阻,患者血液、脑脊液及组织液中5-甲基四氢叶酸不同程度降低,部分患者伴血液甲硫氨酸降低及继发性脑叶酸缺乏症。高同型半胱氨酸血症、低甲硫氨酸血症、脑叶酸缺乏可能是导致神经系统及心脑血管损伤的原因[12]。
MTHFR缺乏症临床表现复杂多样,在新生儿期至成人期均可起病,多系统受累。1972年Mudd等[14]首先报道2例MTHFR缺乏症,血清总同型半胱氨酸浓度均显著增高,1 例以力弱、癫痫发作起病,另1例主要表现为精神分裂症及神经系统症状。早发型于新生儿到1岁内起病,表现为肌张力低下、喂养困难、生长发育停滞、嗜睡、呼吸暂停及小头畸形等[5]。其他症状包括脑室扩张、脑积水及眼震、视神经萎缩等,多无法得到及时诊断及治疗,逐渐加重,发育迟缓,认知障碍,甚至出现癫痫、步态异常、痉挛性瘫痪等神经系统症状。Aljassim 等[1]曾报道7 例MTHFR 缺乏症,其中2 例以脑积水为突出表现,经侧脑室-腹腔分流术及口服药物对症治疗,未得到有效缓解,死于呼吸衰竭。本研究2例患儿均于婴儿期出现脑积水及癫痫,虽然经代谢干预及侧脑室-腹腔分流术治疗,但智力运动发育迟缓及抽搐发作均未得到有效缓解,提示脑积水为MTHFR 缺乏症预后不良的危险因素。晚发型于1岁以后起病,多出现精神行为异常[12,15]、癫痫及共济失调等表现,患者可表现为典型的进展性三相病程,第一相早期发育可正常,第二相出现小头畸形及精神运动发育迟缓,第三相则病情迅速恶化,多因急性呼吸衰竭死亡[16]。患者癫痫可以表现为多种发作形式,如肌阵挛发作、阵挛发作、强直发作等,常见脑萎缩、脑积水、多小脑回畸形及脱髓鞘病变等颅内病变。本研究2 例患儿于婴儿期出现脑积水及癫痫发作,经生化检测及基因分析确诊为MTHFR 缺乏症,与既往报道的病例临床及生化改变相似。
与其他类型的同型半胱氨酸血症不同,MTHFR缺乏症患者多无巨幼细胞性贫血等血液系统异常[16-17]。本研究2 例患儿均未见巨幼细胞性贫血。患者临床症状的严重程度与亚甲基四氢叶酸还原酶的残留酶活性高度相关,体外培养的肝细胞、淋巴细胞、白细胞或成纤维细胞酶活性检测可作为诊断依据[1]。新生儿可通过血液氨基酸分析发现甲硫氨酸及甲硫氨酸/苯丙氨酸比值降低、总同型半胱氨酸水平增高筛查MTHFR缺乏症[4]。
1998 年Goyette 等[18]首次报道了MTHFR基因的结构及定位,MTHFR基因位于染色体1 p 36.22,由12个外显子组成,编码656个氨基酸,迄今已报道100余种致病变异[19],包括错义突变、剪切突变、无义突变等,尚未发现基因型与表型的相关性。最常见的MTHFR致病变异为c.1530G>A,引起9号外显子跳跃,导致移码突变及蛋白质翻译的提前终止[20]。无义突变c.233C>G亦为既往已报道的致病变异[21]。本研究中例2 的c.1530 G>A 为已报道的较常见致病变异,另1 个变异c.233 C >A 与已知致病变异c.233 C>G 的位点及氨基酸改变一致,引起氨基酸翻译提前终止,均可能致病。例1 的2 个新变异c.1915A>C和c.568T>C分别来自父母,在不同物种间存在高度保守性,经致病性预测均可能致病。需要注意的是,MTHFR基因具有大量的多态性变异,c.677C>T最常见,并不是造成严重甲基化障碍的病因[22],因此,应谨慎解读MTHFR基因变异。
目前,对于MTHFR缺乏症尚无根治方法,多数患者通过药物治疗可以改善。确诊后,根据个体情况,应尽早给予甜菜碱、亚叶酸、维生素B12、维生素B6、甲硫氨酸、核黄素、左卡尼汀及对症治疗。其中,甜菜碱〔儿童100~250 mg/(kg·d),成人3~10 g/d〕尤为重要[12],可有效预防神经系统疾病[23]。甜菜碱可通过甜菜碱甲基转移酶作用帮助同型半胱氨酸生成甲硫氨酸,但过量的甜菜碱可能导致甲硫氨酸增高,间接引起脑水肿,所以治疗过程中应密切监测[19]。5-甲基四氢叶酸为唯一能通过血脑屏障的叶酸形式,服用普通叶酸可能会加重大脑中甲基四氢叶酸的缺乏,建议采用亚叶酸或5-甲基四氢叶酸,避免应用叶酸。少数患者可通过补充甲硫氨酸改善临床症状。一氧化二氮可抑制甲硫氨酸的合成,为MTHFR缺乏症的治疗禁忌[19]。
MTHFR 缺乏症患者的严重程度和预后与治疗时间的开始早晚及脑损伤的程度高度相关。通过新生儿筛查发现的患儿经过早期干预,死亡率显著降低,且早期治疗的患儿均未出现显著神经系统后遗症,获得良好预后[23]。若治疗不及时,患儿多因呼吸衰竭或癫痫持续状态夭折。本研究2 例患儿预后不良,与合并脑积水及癫痫等严重神经系统疾病有关。
对于不明原因的智力运动发育障碍、癫痫、精神障碍等患者,需要考虑遗传代谢病的可能,尽早确诊及治疗,改善预后。血液及尿液总同型半胱氨酸测定是发现高同型半胱氨酸血症的关键,血液叶酸、维生素B12、氨基酸及酰基肉碱谱、尿有机酸及基因分析有助于确诊及鉴别诊断。
猜你喜欢
中国生物化学与分子生物学报(2022年7期)2022-09-07
岭南现代临床外科(2022年1期)2022-03-16
中国药科大学学报(2021年6期)2021-12-31
今日农业(2021年16期)2021-11-26
现代畜牧科技(2021年6期)2021-07-16
猪业科学(2018年5期)2018-07-17
中国洗涤用品工业(2015年8期)2015-02-28
无机化学学报(2014年9期)2014-02-28
中国病理生理杂志(2013年10期)2013-01-25
植物营养与肥料学报(2012年2期)2012-10-26
