
一例GATA6基因变异引起儿童特殊类型糖尿病的临床特点及基因变异分析
2024-01-09 02:33应令雯丁宇李娟张倩文常国营郁婷婷王剑祝忠群王秀敏
浙江大学学报(医学版) 2023年6期
应令雯,丁宇,李娟,张倩文,常国营,2,郁婷婷,王剑,祝忠群,王秀敏,2
1.上海交通大学医学院附属上海儿童医学中心内分泌代谢科,上海 200127
2.上海交通大学医学院附属上海儿童医学中心临床研究病区,上海 200127
3.上海交通大学医学院附属上海儿童医学中心遗传分子诊断科,上海 200127
4.上海交通大学医学院附属国际和平妇幼保健院中心实验室,上海 200030
5.上海交通大学医学院附属上海儿童医学中心心胸外科,上海 200127
糖尿病在发病机制和遗传特征上有很大异质性,同时受环境和生活方式的影响,且不同类型的糖尿病中遗传和环境因素的参与度不同。单基因糖尿病是由单一基因缺陷引起的一类分子病因明确的糖尿病,通常呈常染色体显性或隐形方式遗传,部分为母系遗传,存在发病率低、确诊率低和临床异质性高的特点,属于特殊类型糖尿病[1]。根据基因变异位点的功能,其引起糖尿病的潜在机制主要可分为胰腺发育不全或功能不全、胰腺细胞受损以及影响β细胞合成或分泌胰岛素三大类[2],且多合并其他系统疾病,其精准诊治不同于常见的1型糖尿病和2型糖尿病。因此,正确的糖尿病分型诊断对于精准治疗至关重要。
随着高通量测序基因检测技术的发展和普及,越来越多的单基因糖尿病致病基因被发现。其中,以新生儿糖尿病及青少年起病的糖尿病最为常见。然而,仍有许多单基因糖尿病被误诊为1型糖尿病或2型糖尿病。研究表明,45岁以前起病的糖尿病患者中,1%的1型糖尿病和4%~5%的2 型糖尿病最终被证实为单基因糖尿病[3-4]。因此,提高临床医生对单基因糖尿病的认识,患者早期进行基因测序评估是提高本病个体化精准诊治水平的关键。本文报告了1例GATA6基因错义突变引起的特殊类型糖尿病,并结合相关文献进行总结分析,以期提高临床医师对GATA6基因及其引起的特殊类型糖尿病的认识。不同于既往该基因变异常引起的6月龄内发病的新生儿糖尿病,该患儿于1岁后起病,且在确诊前曾发生高渗性高血糖状态,提示先天性心脏病合并高血糖,需密切监测血糖,及时诊治。
1 临床资料
1.1 病历摘要
患儿男性,汉族,2 岁,因“多饮、多食、多尿伴血糖升高两周余”于2018 年11 月30 日收住上海交通大学医学院附属上海儿童医学中心内分泌代谢科。患儿入院时神志清,精神反应可。头围45 cm,坐高38 cm,指间距77 cm,身高81 cm(-2.2 标准差),体重9.8 kg(-2.1 标准差),体重指数14.94 kg/m2(P10~P15)。四肢匀称,无特殊面容及躯体特征。患儿系第2 胎第2 产,足月顺产,出生体重1850 g,身长不详。患儿出生后身高、体重较同龄儿增长缓慢,智力及运动正常。患儿11月龄时因“动脉导管未闭、房间隔缺损Ⅱ型和肺动脉高压”行动脉导管未闭关闭术,术后0~2 d 血糖波动在4.6~8.4 mmol/L,后未规律监测。因患儿术后反复发热,予抗生素、丙种球蛋白、甲泼尼龙等对症支持治疗。术后13 d,患儿出现精神萎靡、叹气样呼吸。实验室检查显示,随机血糖54 mmol/L,糖化血红蛋白15.4%,糖化白蛋白77%,血钠离子156.5 mmol/L,钾离子2.98 mmol/L,有效血浆渗透压372.96 mOsm/L,丙氨酸转氨酶456 U/L,天冬氨酸转氨酶420 U/L,尿素氮27.6 nmol/L,肌酐102 μmol/L,尿酸879 μmol/L,游离三碘甲状腺原氨酸2.42 pmol/L,游离甲状腺素6.08 pmol/L,促甲状腺激素4.12 μIU/mL,提示存在高渗性高血糖状态、肝功能异常、甲状腺功能异常和急性肾功能不全(附表1),予对症支持治疗后均逐步好转,胰岛素治疗6 d 后停用。出院后早期自测血糖正常(空腹血糖4.2~6.1 mmol/L,餐后2 h 血糖5.1~7.6 mmol/L),后未规律监测,术后半年复查糖化血红蛋白为4%,后患者未规律随访。父母为非近亲结婚,患儿有一姐姐,体健,家族成员否认糖尿病家族史。本研究通过上海交通大学医学院附属儿童医学中心伦理委员会审查(SCMCIRB-K2016013),且患儿监护人签署知情同意书。
1.2 辅助检查结果
入院后完善相关检查,糖化血红蛋白14%,糖化白蛋白45%,胰岛自身抗体结果均为阴性,口服葡萄糖耐量试验结果提示患儿空腹和负荷后血糖显著升高(空腹血糖7.6 mmol/L,负荷后2 h 血糖19.4 mmol/L),胰岛素水平低(0.9~2.2 μU/mL),空腹C 肽0.30 ng/mL,负荷后2 h C 肽0.73 ng/mL,较基线C 肽升高1.43 倍,提示仍有部分胰岛功能留存,确诊为糖尿病(类型未定)。患儿肝肾功能、血脂、甲状腺功能正常,腹部超声及CT 检查均未见胰腺形态异常(附表2)。
1.3 基因检测结果及致病性分析
结合患儿为足月小样儿,入院时表现为匀称性矮小,合并先天性心脏病病史,术后出现一过性高渗性高血糖状态,特殊类型糖尿病不能除外。完善全外显子测序结果显示,患儿存在GATA6基因杂合变异c.1366C>T(p.R456C),突变位点位于第4 外显子、第2 个锌指结构域的高度保守区。桑格测序结果显示,其父亲该位点为野生型(图1),母亲未行基因检测。按ACMG 变异分类标准,归类为“可能致病性”变异。
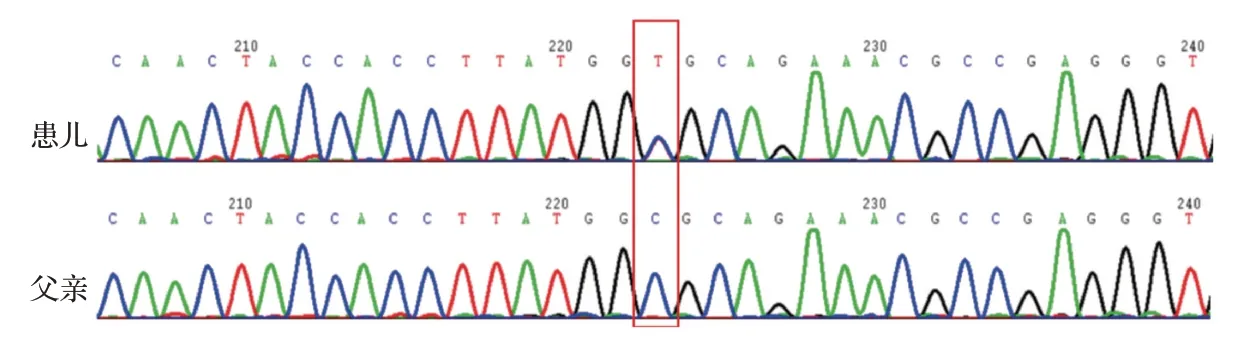
图1 患儿及其父亲GATA6基因测序图Figure 1 Gene sequencing chart of GATA6 of the child and his father
1.4 诊断、治疗及转归
结合其临床表现、辅助检查和基因检测结果,该患儿的糖尿病考虑为GATA6杂合变异所致特殊类型糖尿病,采用短效胰岛素早餐2 IU、中餐1 IU、晚餐1 IU 替代治疗后,血糖波动于6.8~18.1 mmol/L。因长居于外地,患儿出院后未规律随访。末次随访时间为2022 年6 月,距离患儿确诊糖尿病约3.5年(患儿5 岁11 月龄),患儿调整为短效胰岛素早餐3 IU、晚餐3 IU 治疗后,其空腹血糖6.0~10.0 mmol/L,餐后2 h 血糖多数17.0~20.0 mmol/L,偶有血糖超过33.3 mmol/L。
2 讨论
人类GATA6基因位于染色体18q11.2,包含7个外显子,编码595 个氨基酸,主要表达于心脏、胰腺等中、内胚层来源的组织中。现有证据显示,GATA6变异患者的变异位点广泛,并可导致胰腺发育不全、先天性心脏病、消化系统发育障碍、神经认知异常,以及糖尿病、甲状腺功能减退、垂体发育不良、性腺功能减退等内分泌系统疾病,临床表现谱较广[5-9]。然而,其基因型与临床表型无明确相关性[10]。
GATA6蛋白至少包含三个功能结构域,包括1 个转录激活结构域和2 个GATA 锌指结构域(DNA识别和结合所必需)。目前已发现的GATA6变异中,71%发生在功能结构域。其中,大多数错义突变发生在锌指结构域,尤其是第2 个锌指结构域的高度保守区;而导致功能失活的变异多数发生在其转录激活结构域。本文资料中,患儿基因变异位点位于第4 外显子、第2 个锌指结构域的高度保守区,其编码的Arg456残基位于羧基末端锌指的表面,而该位点的变异会破坏GATA6结合和激活靶基因表达的能力,并影响蛋白质之间的相互作用,从而表现为动脉导管未闭、房间隔缺损Ⅱ型、生长发育迟缓和特殊类型糖尿病。
目前已报道5例无亲缘关系的患者存在该位点变异(附表3),因此,c.1366C>T(p.R456C)变异是迄今GATA6中同时影响心血管系统及内分泌系统中最常见的变异。Allen等[11]于2011 年首次报道了该位点变异的2 例患者,其中一例表现为胰腺发育不全、新生儿糖尿病、室间隔缺损、永存动脉干、癫痫发作和发育迟缓,另一例则表现为胰腺发育不全、新生儿糖尿病、法洛四联症、先天性膈疝和发育迟缓。Yu等[12]报道了该位点变异的患儿主要表现为法洛四联症、单脐动脉和先天性膈疝。Sanyoura等[13]报道了该位点变异的1 例患儿表现为青春期糖尿病、高血压、高脂血症、甲状腺功能减退和先天性心脏病(4A 型动脉干、主动脉弓中断、动脉干瓣膜关闭不全和室间隔缺损)。Raghuram等[14]报道了该位点变异的1 例患儿表现为胰腺发育不全、新生儿糖尿病、胆道闭锁、永存动脉干、甲状腺发育异常、可能的肾上腺功能不全、血小板减少症和新生儿中风。本文资料中的患儿主要表现为先天性心脏病(动脉导管未闭和房间隔缺损)和糖尿病,伴生长发育迟缓,并在先天性心脏病术后出现了一过性高渗性高血糖状态,病程中尚有肝功能异常、甲状腺功能异常和急性肾功能不全。考虑到该患儿后续复查肝肾功能、甲状腺功能未见异常,其术后出现的暂时性肝功能、急性肾功能不全及甲状腺病态综合征或与手术应激有关。由此可见,尽管是同一位点变异,其具体临床表现仍有明显的差异,但先天性心脏病和血糖异常仍是该位点变异的主要表现。
高渗性高血糖状态是糖尿病严重的急性并发症之一,以血糖、血浆有效渗透压显著升高而无明显酮症为特征[15],多见于2 型糖尿病老年患者,在儿童和年轻人群中罕有报道[16]。然而,儿童高渗性高血糖状态的发病率可能在逐步增加[17]。研究表明,停用降糖药物、感染等导致的应激反应可能是诱发高渗性高血糖状态的重要原因,而脱水的严重程度以及是否有合并症、高龄是影响患者预后的主要因素[15,18-19]。
本研究提示GATA6基因变异与高渗性高血糖状态可能相关。目前认为GATA6变异是导致胰腺发育不全的主要原因之一,同时,GATA6 可能参与了胰岛素的合成与分泌过程[11,20]。现有证据显示,GATA6变异引起的糖代谢异常临床表现多样。具体而言,GATA6杂合变异主要表现为胰腺完全或部分缺如,大多功能完全丧失,从而导致患儿表现为宫内发育迟缓、新生儿糖尿病和胰腺外分泌功能衰竭;此外,也有部分患者临床表现轻微,仅有胰腺细胞或胰岛β细胞数的减少,从而表现为青少年或成年起病的糖尿病[5,21]。对于本例患儿,尽管未出现胰腺发育不全,且术前血糖水平正常,但GATA6基因变异可能使其胰岛β 细胞数减少,并影响其胰岛素的合成及分泌过程。该患儿经历先天性心脏病手术及感染引起应激反应、发热引起不显性失水、糖皮质激素的应用等综合因素,血糖水平短期内急剧升高,从而导致高渗性高血糖状态的发生。对于患儿高渗性高血糖状态时明显升高的糖化血红蛋白及糖化白蛋白考虑系血糖很高时高渗状态下的“检测假象”。然而,并未及时在患儿高渗性高血糖状态纠正后再次评估糖化血红蛋白及糖化白蛋白或是导致患儿未能早期明确诊断的原因之一。因此,对于应激后出现高渗性高血糖状态、酮症酸中毒等糖尿病急性并发症且伴有先天性心脏病的患儿,需评估是否有GATA6基因变异的可能。
本研究尚有一定局限性。尽管患儿母亲及其姐姐自幼体健,无先天性心脏病或糖尿病相关病史,然而无法获得患儿母亲测序结果以明确患儿的致病基因来源仍是本研究的不足之处。与此同时,患儿为足月小样儿,而既往研究指出足月小样儿由于生长追赶容易伴发胰岛素抵抗。本例患儿术前仅评估了其血糖水平,未对患儿进行胰岛功能筛查。
综上,本文报道了1例GATA6杂合变异所致的特殊类型糖尿病患儿,该患儿在病程中曾因应激出现一过性高渗性高血糖状态,提示GATA6基因杂合变异的临床表型异质性并非都在新生儿阶段起病,也可表现为一过性血糖异常。对于同时患有先天性心脏病和胰腺发育异常或血糖异常的幼儿,应完善临床检查程序并在此基础上早期筛查GATA6基因以明确致病原因,从而为遗传咨询及后续治疗提供依据。同时,对该类患儿需持续警惕糖尿病的发生,密切监测血糖,及早干预,避免不良临床结局。
本文附表见电子版。

志谢研究得到国家自然科学基金(81900722,82170910)支持
AcknowledgementsThis work was supported by the National Natural Science Foundation of China (81900722,82170910)
利益冲突所有作者均声明不存在利益冲突
Conflict of InterestsThe authors declare that there is no conflict of interests
©The author(s) 2023.This is an open access article under the CC BY-NC-ND 4.0 License (https://creativecommons.org/licenses/by-nc-nd/4.0/)
猜你喜欢
金秋(2021年18期)2021-12-02
自我保健(2021年4期)2021-06-16
中国保健营养(2019年6期)2019-10-21
广州大学学报(自然科学版)(2019年1期)2019-05-07
中西医结合肝病杂志(2018年2期)2018-09-12
天津科技大学学报(2016年1期)2016-02-28
中国卫生标准管理(2015年15期)2016-01-15
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
中国医学装备(2015年10期)2015-12-29
中国当代医药(2015年23期)2015-03-01
