
Harel-Yoon综合征一家系临床表型及ATAD3A基因变异分析
2024-01-09 02:33郑祎于欣雨张婷胡凌微周朵黄新文
浙江大学学报(医学版) 2023年6期
郑祎,于欣雨,张婷,胡凌微,周朵,黄新文
1.浙江大学医学院附属儿童医院遗传与代谢科,国家儿童健康与疾病临床医学研究中心,浙江 杭州 310052
2.浙江大学医学院附属第一医院肝胆胰外科,浙江 杭州 310003
ATAD3A基因位于染色体1p36.33,编码线粒体膜蛋白,对维持线粒体功能至关重要[1-2]。该基因变异可引起极为罕见的常染色体隐性或显性遗传病,临床表现异质性大,可有外观畸形、高乳酸血症、肥厚型心肌病、脑桥小脑发育不全等症状。截至目前,文献报道ATAD3A基因变异80例,临床表型包括HAYOS、PHIRNL、遗传性痉挛性截瘫等[2-4]。本文通过全外显子组测序确诊一个ATAD3A基因变异家系,探究其临床表现及基因特点,为遗传学诊断和咨询提供依据。本研究对象或其家属签署知情同意书,研究方案通过浙江大学医学院附属儿童医院医学伦理委员会审查(2021-IRB-292)。
1 临床资料
1.1 病历摘要
先证者女性,11 d,因“咳嗽伴口吐白沫3 d”入住浙江大学医学院附属儿童医院。先证者系孕39+2周单胎顺产娩出(孕2 产2),出生体重3.17 kg,出生时Apgar 评分正常,否认宫内窒息史和窒息抢救史,父母亲否认近亲婚配及家族遗传病史。入院时体重3.14 kg,身长50 cm,体温36.5 ℃,心 率198 次/min,呼 吸62 次/min,血 压63/32 mmHg(1 mmHg=0.133 kPa)。患儿神志清,精神尚可,皮下脂肪薄,追声追视较差;心率快,心音弱,呼吸急促,未闻及明显杂音;四肢肌张力增高,上肢为主。实验室检查结果显示,白细胞计数12.3×109/L,中性粒细胞百分比30.5%,超敏C 反应蛋白0.54 mg/L;心肌损伤标志物升高:氨基末端脑利钠肽前体28 037.7 pg/mL(参考值<300.0 pg/mL),肌红蛋白101.5 ng/mL(参考值2.75~31.03 ng/mL),肌酸激酶同工酶8.9 ng/mL(参考值<5.0 ng/mL),特异性肌钙蛋白0.25 ng/mL(参考值<0.40 ng/mL);血浆乳酸4.0 mmol/L(参考值0.5~1.6 mmol/L),pH 值小于7.00;血串联质谱酰基肉碱检测辛酰基肉碱、癸酰基肉碱偏高,分别为0.45 μmol/L(参考值0.02~0.17 μmol/L)、0.51 μmol/L(参考值0.03~0.22 μmol/L)。心脏超声检查提示肥厚型心肌病,左、右心室收缩力减小,房间隔缺损,卵圆孔未闭,射血分数32%~69%。心脏磁共振检查提示左心室心肌不均匀增厚伴异常信号,伴少许纤维化。心电图检查提示T波改变及高侧壁ST段改变。胸部X线检查提示右下肺可见片状密度增高影。头颅磁共振检查提示双侧大脑半球脑白质及脑桥T2信号偏高,额颞部脑外间隙稍宽。脑电图检查正常。患儿住院3个月余,反复出现心力衰竭、高乳酸血症、喂养困难、体重不增,最终于3.5月龄时死于心肺功能衰竭。
先证者姐姐足月顺产娩出,出生时Apgar 评分正常,于18 d因“少吃少动2 d,反应差1 d”入住浙江大学医学院附属儿童医院。当时昏迷状态,肌张力减小,血气提示代谢性酸中毒,pH 值小于7.00,乳酸24.0~29.0 mmo1/L;心肌损伤标志物升高:乳酸脱氢酶1954 U/L,肌酸激酶1026 U/L,肌酸激酶同工酶活性539 U/L;胸部X线检查提示两肺渗出性改变,心影大。患儿当日即死于“新生儿败血症、心功能不全”。
1.2 基因检测结果及致病性分析
分别采集先证者和父母2 mL EDTA抗凝外周血,提取基因组DNA,利用序列捕获技术(Agilent SureSelect 系列试剂盒)将全基因组外显子区域DNA 捕获并富集后进行PE150 高通量测序(美国Illumina 公司),结果显示,先证者ATAD3A基因chr1:1463113(NM_001170535.3)Exon:14 和chr1:1463228(NM_001170535.3)Exon:14 存在复合杂合变异,为基因移码突变c.1492dup(p.T498Nfs*13)和错义突变c.1376T>C(p.F459S),分别遗传自其父亲和母亲(图1)。将先证者姐姐的新生儿筛查干血斑进行基因检测(先证者父母提供),结果显示其ATAD3A基因存在与先证者相同的变异。

图1 先证者及其父母基因组DNA高通量测序结果Figure 1 High-throughput sequencing results of the proband and their parents
检索GnomAD、1000 Genomes、ClinVar、LOVD 和HGMD数据库,患儿两个变异位点均未见报道。根据美国医学遗传学和基因组学指南,c.1376T>C(p.F459S)为错义突变,用SIFT、PolyPhen、FATHMM、MutationTaster软件预测均为有害突变,变异评估为“临床意义未明”;c.1492dup(p.T498Nfs*13)为移码突变,预测可导致多肽链合成提前终止,变异评估为“致病”。
将ATAD3A 氨基酸序列提交到i-TASSER 预测其三维结构,使用PYMOL 软件对新发错义突变的结构稳定性进行分析[5]。结果显示,野生型459 绿色位点的苯丙氨酸在被丝氨酸取代后,该区域苯基与周围氨基酸产生的疏水键变少,与周围原子产生的次级键变少,不利于维持蛋白质的三级结构,蛋白质稳定性下降(图2)。
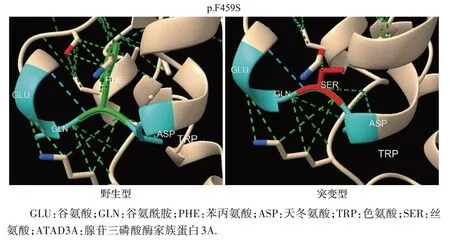
图2 ATAD3A部分三级结构p.F459S变异的结构稳定性分析Figure 2 Structural stability analysis of the p.F459S variant of the partial tertiary structure of the ATAD3A protein
2 讨论
ATAD3A定位于染色体1p36.33,含16 个外显子,大小约22.5 kb,在人类中,ATAD3A复制两次形成ATAD3A、ATAD3B和ATAD3C串联排列的ATAD3基因簇[6]。ATAD3A在胚胎早期至成年期的所有组织中普遍表达,ATAD3B对ATAD3A起负调控作用,仅在人类胚胎干细胞中特异性表达,而ATAD3C可能是假基因[7]。三个高度同源的旁系同源阵列的遗传结构使该区域易发生基因重排[1]。ATAD3A编码的含有ATP 酶家族AAA 结构域的蛋白3 成员A 是一种线粒体膜蛋白,其羧基末端面向基质形成六聚体环状结构,氨基末端区域位于外膜与其他酶复合,参与线粒体嵴结构形成以及与内质网的连接交换,在线粒体生物发生、线粒体自噬、mtDNA 维持和胆固醇代谢中发挥重要作用[3,8-9]。ATAD3A缺失会导致小鼠、黑腹果蝇和秀丽隐杆线虫的早期胚胎或幼虫死亡,提示ATAD3A在胚胎发生中具有关键作用[4,10]。
ATAD3A基因变异导致的遗传病首次由Harel等[4]在2016 年报道,截至2023 年11 月共检索到80 例患者[1-4,11-24],来自61 个家系,男女比例为45∶35,发病年龄为宫内26 周至儿童期,死亡52例(65.0%),死亡年龄中位数为1 个月(宫内36+6周~2 岁),最大随访年龄40 岁。主要临床表现有外观畸形(61.3%)、角膜浑浊或先天性白内障(57.5%)、高乳酸血症(57.5%)、肌张力减小(55.0%)、肥厚型心肌病(46.3%)、小脑萎缩或发育不全等中枢神经系统异常(50.0%),见附表1。其他表现包括代谢障碍(甲基戊二酸尿症、三羧酸循环中间产物增加),癫痫、急性脑病、多发性挛缩等神经系统表现,视神经萎缩、视力下降、畏光、眼球震颤等眼部表现及胎儿水肿、肺泡发育不全、肝肿大等。极少数患者有听力丧失、注意力缺陷多动障碍、牙釉质缺陷、桥本甲状腺炎、硬皮病等。
ATAD3A变异引起临床表型主要有三类。PHIRNL 为常染色体隐性遗传,常在宫内发病并在新生儿期死亡,临床表现严重,常有角膜混浊、癫痫发作、畸形面容和进行性脑桥小脑发育不全、呼吸功能不全症状,需要机械通气和重症监护[7,12]。ATAD3A变异导致的遗传性痉挛性截瘫为常染色体显性遗传,主要表现为双下肢或四肢痉挛性瘫痪及畏光、视力下降症状[11]。HAYOS为常染色体隐性或显性遗传,临床表现差异大,核心特征为发育迟缓、肌张力减退、痉挛、周围神经病变和肥厚型心肌病,常伴有高前额、三角面容、小颌畸形、低位耳、鸡胸、足部畸形等外观异常及智力缺陷;常见血浆和(或)脑脊液乳酸增多,头颅磁共振光谱乳酸峰升高,提示小脑萎缩或发育不全[4]。本文先证者及其姐姐于新生儿早期出现体格发育迟缓、难以纠正的高乳酸血症、代谢性酸中毒、肥厚型心肌病及肌张力减退,符合HAYOS诊断。
截至目前,报道的80例患者涉及的ATAD3A基因变异包括错义突变(36.27%)、重复突变(21.57%)、缺失突变(21.57%)、移码突变(8.82%)、剪接突变(6.86%)、无义突变(2.94%)和非编码区突变(1.96%)7种变异方式(附表2)。其中缺失突变中有4 例患者双等位基因ATAD3B/ATAD3A缺失(3.92%),未报道具体变异位点。基因变异类型与临床表型具有一定相关性。大片段缺失变异为隐性遗传,由于两条同源染色体的ATAD3A基因均缺失,细胞中ATAD3A表达显著降低,引起mtDNA聚集、胆固醇代谢异常,严重影响发育,绝大部分患儿于新生儿期死亡,磁共振提示小脑或脑干发育不全(85.7%)等[2,4],表型为PHIRNL。大片段重复突变为显性遗传,源于非等位基因同源重组产生ATAD3A-C融合基因,ATAD3A中ATP酶结构域高度保守的精氨酸指(Arg466)在ATAD3A-C 融合蛋白中变为半胱氨酸,可能会抑制ATP 酶的活性,细胞中90%的ATAD3 六聚体掺入了ATAD3A-C 融合蛋白[3,25]。患者临床常表现为特征性的致命性围产期心肌病(77.3%)、持续性高乳酸血症(86.4%),脑桥小脑发育不全与周围神经症状较轻的HAYOS,其中g.1391996_1460043dup变异共报道12例,是截至目前报道最多的变异位点(11.8%)[17]。ATAD3A显性错义突变可能通过上调Ⅰ型干扰素的途径发挥致病作用,表现出较轻的小脑发育不全和更广泛的神经和多系统疾病,且常伴有自身免疫疾病[18]。其中c.1064G>A(p.Gly355Asp)错义突变位于ATP 酶结构域的ATP 结合Walker A 子域,临床表型为遗传性痉挛性截瘫[2,4,11]。ATAD3A复合杂合无义突变目前仅报道2例,表现为明显的外观畸形、小脑脑干萎缩或发育不全、角膜混浊,分别于出生后6 和3 d 死亡,临床表型为PHIRNL[12]。移码突变和错义突变复合杂合变异共报道3个家系6 例患者[1,15],移码突变位于ATP 酶结构域中,而错义突变位于ATP 酶结构域外。6例患者随访期间均存活,其中5 例年龄在17 岁及以上,临床表现为发育迟缓、周围神经系统病变、肌张力减低、下肢痉挛、先天性白内障、视力下降等。本文先证者及其姐姐为移码突变和错义突变复合杂合变异。但这两个变异均位于ATP 酶结构域,对ATAD3 表达及活性影响较大,结构预测有害,导致本家系的两例患者发病时间早,肥厚型心肌病和高乳酸血症症状突出,生存时间短暂。
目前,ATAD3A基因变异尚无有效治疗方法,以对症支持治疗为主。生酮饮食可能通过改善线粒体功能、促进线粒体生成延缓患者小脑萎缩进展[21,26]。该基因变异患者预后差,随访期间死亡52例(66.7%),中位数年龄1 月龄,致死性的新生儿脑桥小脑发育不全是重要的死亡原因,存活患者往往遗留神经系统症状。
综上所述,本文报道了一个ATAD3A基因复合杂合变异导致HAYOS 的家系两例病例,第一例突发死亡没有明确遗传学病因,导致第二胎又出现同样病例,因此对于早发不明原因心功能不全、代谢性酸中毒、高乳酸血症、小脑或脑桥发育不全、肌张力低下的患儿,即使死亡的患儿,应尽早完善基因检测明确遗传学病因,指导产前诊断以避免同样病例再次出生。
本文附表见电子版。

志谢研究得到国家自然科学基金(82073560)支持
AcknowledgementsThis work was supported by the National Natural Science Foundation of China (82073560)
利益冲突所有作者均声明不存在利益冲突
Conflict of InterestsThe authors declare that there is no conflict of interests
©The author(s) 2023.This is an open access article under the CC BY-NC-ND 4.0 License (https://creativecommons.org/licenses/by-nc-nd/4.0/)
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30
临床输血与检验(2022年3期)2022-06-22
昆明医科大学学报(2022年3期)2022-04-19
世界最新医学信息文摘(2021年12期)2021-06-09
中南医学科学杂志(2019年6期)2019-12-05
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
家庭医药(2018年6期)2018-06-25
临床超声医学杂志(2017年9期)2017-09-26
磁共振成像(2015年2期)2015-12-23
