
淡水鱼类环境DNA宏条形码引物的筛选及其在千岛湖的应用*
2024-01-13 07:41胡文静李志力刘其根胡忠军
湖泊科学 2024年1期
周 严,童 璐,胡文静,李志力,郝 雷,刘其根,胡忠军**
(1:上海海洋大学,农业农村部鱼类营养与环境生态研究中心,上海 201306) (2:上海海洋大学,农业农村部淡水水产种质资源重点实验室,上海 201306)
维持生物多样性的稳定是生态系统可持续发展的基础,由于人类的过度开发、气候变化、环境污染、外来物种入侵等因素,全球野生动物资源量正在大幅缩减,据2022年世界自然基金会发布的地球生命力报告显示,自1970-2018年之间,全球野生动物种群数量消亡了69%,地球生命力指数下降幅度超过一半以上[1]。截至目前,全球共有3万多种鱼类,占所有脊椎动物的一半以上,具有重要的生态效益和经济价值[2-3]。随着对鱼类受威胁状况了解的深入,鱼类所受威胁的认知度也不断加深,以中国为例,在最新的《中国生物多样性红色名录》(脊椎动物卷)评估中受威胁的鱼类比例高达22.5%,鱼类保护工作的实施已迫在眉睫[4]。鱼类多样性的有效保护依赖于对现有鱼类群落生态和动态的深入了解,这需要对鱼类的群落结构进行全面和准确的评估。传统的鱼类多样性评估多依靠捕捞如电捕、网捕等方式进行样品的采集,不仅需要大量的人力和时间,而且成本也较高;同时,基于形态学的物种鉴定非常容易出错,处在生活史早期阶段的鱼类鉴定尤为如此,因此鱼类样品的准确分类需要依赖专业的鉴定人员[5]。此外,基于捕获的调查方式往往会对鱼类本身造成伤害,反而使得鱼类的资源量变得更少,这也违背了生物多样性保护工作的初衷[6]。
eDNA(environmental DNA)是指从环境样品(如土壤、水或空气)中提取的片段DNA,不需要预先捕获任何目标生物[7]。eDNA方法是基于直接从环境样本提取到的生物体DNA进行序列分析,可以在不捕获实际生物体的情况下对目标物种进行识别[8]。自2008年Ficetola首次成功将eDNA技术应用于水生生物调查以来,越来越多的人开始使用这种方法,基于eDNA的生物多样性监测已被证明是一种高效、经济和无创的生物监测方法[8-10]。将宏条形码引物与高通量测序相结合,eDNA方法在鱼类多样性监测方面取得了巨大的进步,与传统的调查方式相比较,其能够更好地揭示物种的丰富度,已彻底改变了动植物资源的调查方式[11-13]。
目前,基于eDNA的生物多样性监测技术仍不够完善,需进一步开展系统的研究和验证[14]。在技术应用方面,宏条形码引物合适与否常被认为是影响eDNA监测结果准确性的主要因素之一[15]。不同引物对于鱼类物种的覆盖率和分辨能力有所不同,尤其是当引物对于鱼类环境DNA的扩增效率比较低、存在许多非目标物种的扩增,或对不同物种的序列区分能力不足时,往往会出现假阳性或者假阴性的鉴定结果,而理想的引物应具有较高的物种覆盖率、较强的物种分辨率、适当的条形码长度以及更少的非特异性扩增等优势[15-16]。因此,引物的开发或筛选是提高eDNA监测效率和可靠性的关键步骤[13,17]。
过去有研究者针对鱼类群落开发了部分宏条形码引物,并且成功地描述了当地的鱼类多样性[18-19]。由于很难对这些来自不同研究中的宏条形码引物的监测效果进行比较与评估,许多研究人员仅依据个人的主观判断从众多引物中挑选一对引物对鱼类物种多样性展开研究,并未深入分析该引物的特征以及其对所研究群落的适用性[20-21]。近年来,相关研究开展了eDNA宏条形码引物的分析和测试,但仅评估了少量的引物[22],也有部分研究选取较多的引物开展了相对全面的评估[16,23],伴随新的引物不断被开发出来[24-25],与之前常用的宏条形码引物相比,新开发出来的引物是否有更好的表现,仍需进一步验证。因此,有必要对现有的宏条形码引物进行全面评估,并探讨它们在基于eDNA技术的鱼类物种监测中的适用性和可靠性。
本研究结合计算机模拟PCR和野外样品实验验证,比较评估已发表的基于12S、16S、COI和Cytb线粒体基因设计的27对引物,以及自行设计的2对引物,共29对引物。首先,计算机模拟PCR分析来自Mitofish数据库鱼类基因数据的物种覆盖率和扩增长度等,接着对来自千岛湖五大湖区及其支流的eDNA样品进行宏条形码分析以综合评价不同宏条形码引物优劣势,为今后基于eDNA技术的鱼类物种多样性评估提供参考。
1 材料和方法
1.1 引物的检索与设计
为尽可能全面地搜集有关鱼类的宏条形码引物,在科学网(www.webofscience.com)以eDNA或environmental DNA或eDNA metabarcoding为关键词进行文献检索,共检索到27对已应用于eDNA研究且效果较好的宏条形码引物,包括了12S rRNA、16S rRNA、COI和Cytb基因。同时,基于千岛湖历史记录中的鱼类物种,从NCBI(https://www.ncbi.nlm.nih.gov/)和Mitofish数据库(http://mitofish.aori.u-tokyo.ac.jp/)中下载了105种鱼类的127条线粒体DNA序列,使用Primer Premier 5.0软件[26]基于12S rRNA区域设计了2对宏条形码引物,以上所得到的eDNA宏条形码引物汇总如表1所示。

表1 本研究分析的22对鱼类宏条形码引物Tab.1 Summary of the 22 fish metabarcoding primer sets analyzed in this study
1.2 计算机模拟PCR
于2021年11月,从Mitofish数据库中下载3118种鱼类的线粒体DNA序列,构建了标准鱼类线粒体DNA条形码数据库。首先,使用PrimerMiner对Mitofish数据库中3118种鱼类进行评估,以此来计算宏条形码引物对于Mitofish数据库中不同鱼类物种的覆盖率和分辨率[40],允许的最大错配数为3 bp。随后使用Tbtools对成功扩增的序列长度进行分析[41]。最后通过megaX计算扩增序列之间的平均遗传距离[42],以评估不同宏条形码引物所扩增鱼类序列之间的差异程度。当引物扩增的物种数目大于所有引物扩增鱼类物种数的平均值且平均扩增长度处于100~500 bp之间时,该引物被用于下一步实验,以进一步评估其对于实际环境DNA样本中鱼类的监测能力。
1.3 eDNA样品的采集与保存
千岛湖位于浙江省杭州市淳安县境内,有着良好的生态环境和丰富的鱼类资源,自20世纪以来的历史标本和网捕调查结果记录了上百种鱼类[43]。为了评估引物对于实际采集的环境DNA中鱼类的监测效果,本研究对来自千岛湖湖区及其支流的水环境DNA进行了验证。在2020年12月-2021年10月,从千岛湖五大湖区和14条支流共计39个站点进行了水样的采集。通过玻璃采水器,在每个采样点水下约0.5、5和10 m的位置收集3个2 L的水样,将水样混合均匀后放置在冰上运回上海海洋大学千岛湖基地实验室,在12 h内全部过滤。抽滤过程中所涉及的镊子、过滤器均提前置于10%的次氯酸钠溶液中浸泡10 min,并使用纯水冲洗干净。每次过滤前均设置2 L无菌水作为阴性对照,使用孔径0.22 μm的混合纤维素滤膜(生工生物)对采集的水样进行过滤,过滤完后将滤膜对折,放置于2 mL离心管中,采用干冰冷冻保存。
1.4 传统鱼类资源调查
于2020年9月-2021年11月对千岛湖五大湖区及其主要支流开展了野外调查,在千岛湖湖区及河湖交汇处的淌水区表层、5 m水层、10 m水层分别放置多网目单层刺网1套,在沿岸带放置多网目单层刺网1套、定置串联倒须笼网2个。支流主要依靠定置串联倒须笼网的方式进行鱼类多样性调查,在每个采样点放置定置串联倒须笼网2个。傍晚日落前1 h左右放网,其在水中浸泡约12 h后,次日清晨收网。收集所有鱼类,用低温保温箱将鱼类运回上海海洋大学千岛湖基地实验室,进行物种鉴定和形态学数据的测量。
1.5 eDNA的提取、PCR扩增和测序
样品运输至实验室后,采用Fast DNA Spin Kit(MPBio)试剂盒并参照说明书进行总DNA的提取。提取后的DNA经1%琼脂糖凝胶电泳检测,并用NanoDrop 2000质检合格后于-20℃保存备用。对于计算机模拟PCR中扩增长度在100~500 bp且扩增出的物种数在所有引物扩增鱼类物种数的平均值之上的引物进行下一步实验,以原始设计论文中使用的Tm值为中心,在10~20℃范围内的不同温度梯度下,使用通用程序执行8~12个重复PCR,确定每个引物组的最佳退火温度。针对每一对引物,均进行至少两次梯度PCR实验,以确保扩增效果的一致性。对于每个引物组,在明确最适退火温度后,使用已提取好的21个eDNA样本和3个阴性对照进行PCR扩增,每一次PCR均为3个重复,以确保PCR产物的量足以进行测序。每次PCR反应的总体积为25 μL,其中模版DNA 1 μL、正反引物各1 μL(10 μmol)、Ultra HiFidelity PCR Kit II(天根生物)12.5 μL,ddH2O 9.5 μL。PCR反应程序为:98℃ 30 s;98℃ 10 s,退火温度(Tm)20 s,72℃ 20 s,35个循环;72℃ 5 min。随后通过1%琼脂糖凝胶电泳对PCR产物进行检测,确保检测结果均为一条单一明亮的条带,将每一条带进行切胶,纯化回收,并用NanoDrop 2000分光光度计(Thermo Scientific)测定DNA浓度,将每组引物扩增纯化后的21个PCR产物进行等量混合,利用Illumina公司的Miseq PE300平台进行测序,进一步的文库构建和上机测序由上海美吉生物技术有限公司进行。
1.6 测序数据的生物信息学处理
基于fastp[44](https://github.com/OpenGene/fastp,version 0.20.0)软件去除引物接头序列,并对原始测序序列进行质控,过滤掉低质量的Reads。采用FLASH[45](http://www.cbcb.umd.edu/software/flash,version 1.2.7)软件根据Reads间的重叠关系,对严格过滤后的Reads进行拼接。使用软件USEARCH(v7.0.1090)根据97%的相似度将序列聚类为可操作分类单元(OTU)[46]。为了解每个OTU所对应的物种,采用BLAST算法对OTU代表序列进行分类学比对,参考数据库为Nt数据库(https://www.ncbi.nlm.nih.gov/),将序列一致性≥97%作为阈值,对OTU代表序列进行注释。使用Qiime软件基于bray_curtis距离算法计算beta多样性距离矩阵[47],使用R语言(version 3.3.1)vegan软件包进行NMDS分析和作图[48]。
2 结果
2.1 计算机模拟PCR分析
本研究通过计算机模拟PCR对来自Mitofish数据库中的3118种鱼类序列进行扩增,不同宏条形码引物对于不同鱼类物种的覆盖度和分辨率分析结果如图1所示。使用29对引物所扩增出的平均鱼类物种数为2523种,引物12S-V52扩增出的鱼类物种数最多,一共扩增出了3113种鱼类,引物GFCC扩增的鱼类物种数最少,仅扩增出了429种鱼类。对扩增出的鱼类物种作进一步分析,平均每对引物能够准确鉴定的物种数为2144种,引物16SAR/BR能够准确鉴定的鱼类物种数最多,为2782种,引物GFCC能够准确鉴定的物种数最少,为336种。

图1 不同引物能够扩增出的物种数和能够明确鉴定出的鱼类物种数量Fig.1 Number of species that can be amplified and number of fish species that can be unambiguously identified by different primers
通过对不同宏条形码引物扩增出的鱼类序列长度作进一步分析,大多数宏条形码引物的扩增长度处于100~500 bp之间。不同eDNA宏条形码引物扩增的平均条形码长度为305 bp,其中FishF1/R1扩增出了最长的长度,为655 bp,引物Teleo扩增出了最短的长度,为63 bp(图2)。

图2 基于计算机模拟PCR结果计算出的每对引物平均扩增长度(虚线表示用于下一步实验验证时扩增片段长度的阈值)Fig.2 Average amplification length per primer pair calculated based in silico PCR results
2.2 遗传距离分析
基于不同引物扩增出的鱼类物种,计算了不同物种之间的平均遗传距离。29对引物扩增的鱼类物种间遗传距离的平均值为19.02%±3.52%(图3),引物ChordB扩增出的鱼类序列之间平均遗传距离最大,其次是Fish16S和16SFishS,而引物L2513扩增出的鱼类序列之间平均遗传距离最小。
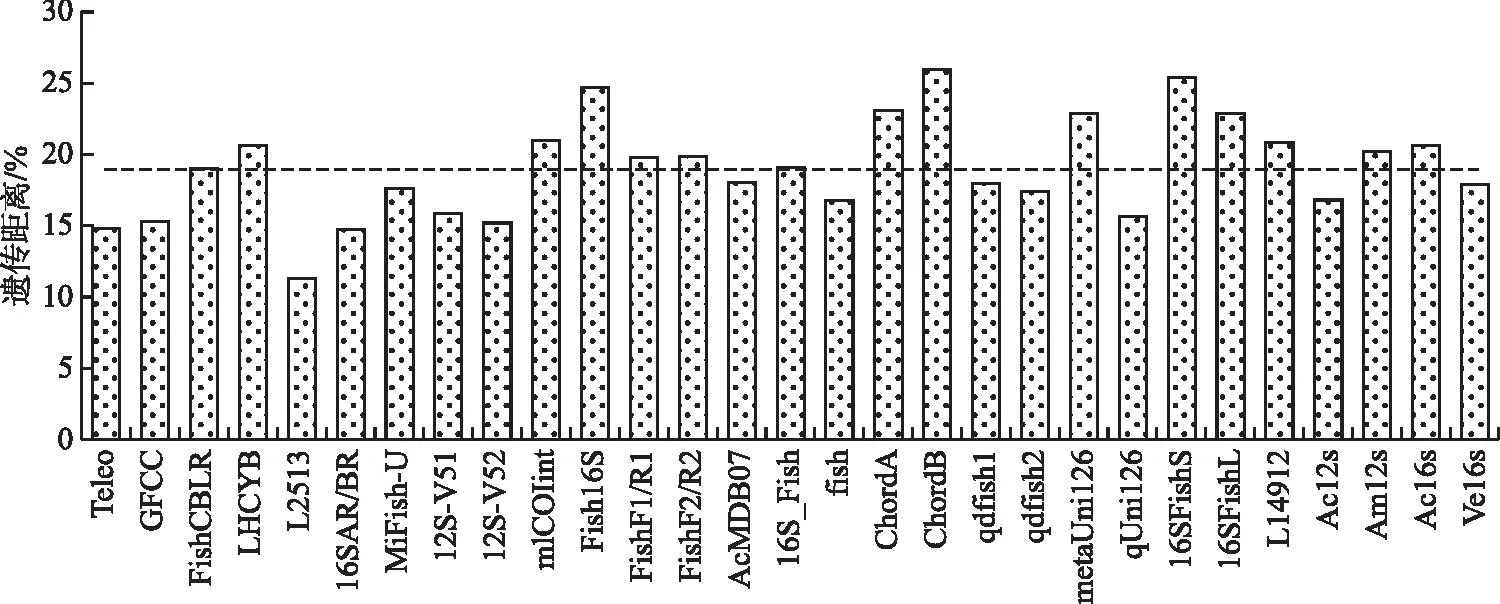
图3 不同引物扩增出的鱼类序列之间的遗传距离(虚线表示平均值)Fig.3 Genetic distance between fish sequences amplified by different primers
2.3 高通量测序结果
基于计算机模拟PCR的结果,有17对引物成功扩增的物种数以及扩增片段长度处于符合的范围内而被用于下一步分析。通过对已提取的eDNA样本进行PCR扩增和上机测序,经过质控、拼接、聚类以及数据库比对,17对引物中有15对被成功扩增并得到了与之相关的测序结果,另外2对引物由于扩增的序列没有匹配到具有≥97%相似性的鱼类序列而被删去。15对引物所构建的测序文库共获得有效拼接序列961740条,有效拼接序列最多的为Ac16s,共获得127385条序列,最少的为Ac12s,共获得31255条序列。从物种稀释曲线图(图4)可知,随着测序量的增加,曲线逐渐趋向于平缓,由此可以看出测序深度足以覆盖样品中所有的物种,可以用于后续分析。
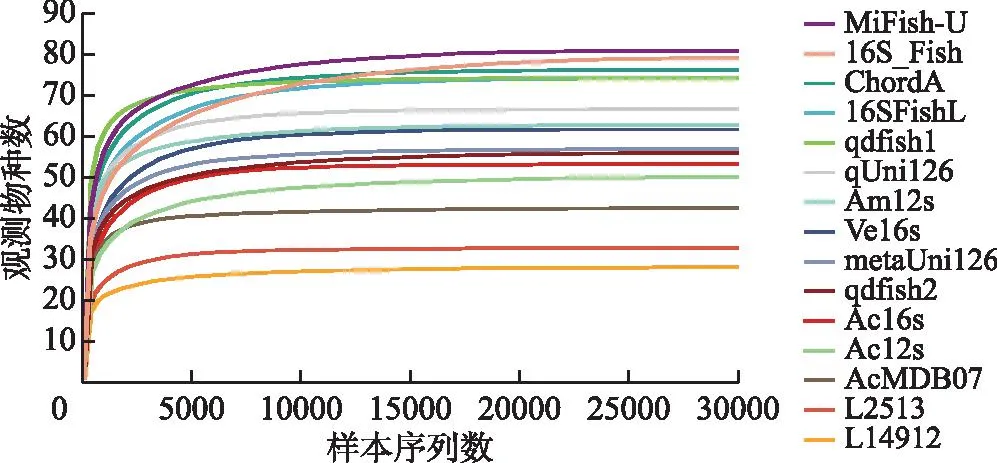
图4 使用不同引物所得到的物种稀释曲线Fig.4 Rarefaction curves of the observed species in different primers
2.4 鱼类物种组成
不同宏条形码引物所监测到的鱼类物种数量如图5所示,平均每对引物监测出的鱼类物种数为60种,引物MiFish-U、16S_Fish、ChordA、16SFishL、qdfish1监测到的鱼类物种数相对较多,而其他引物监测到的物种数相对较少(如L14912只扩增出了28种鱼类)。将不同宏条形码引物监测到的鱼类物种与传统鱼类资源调查结果进行对比,尽管监测到的物种数排名前四的引物仍为MiFish-U、16S_Fish、ChordA、16SFishL,但监测到物种数最多的宏条形码引物是ChordA(58种),其次是16SFishL(57种),然后是引物16S_Fish和MiFish-U。
基于不同的宏条形码引物所获得测序结果,选取丰度排名前50的鱼类物种,绘制了鱼类物种组成差异图。如图6所示,丰度排名前50的鱼类中,16SFishL、ChordA、Mifish-U、qdfish1监测的物种数最多,均监测到了40种以上的鱼类;而引物L14912和L2513监测到的物种数最少,仅为19种。不同的引物既显示出了各自所具备的特性也显示出了不足之处,一方面,本研究中所用到的引物对于大部分鱼类物种均能够监测到,另一方面,也有一些物种仅仅只被一对引物或个别引物所监测到。
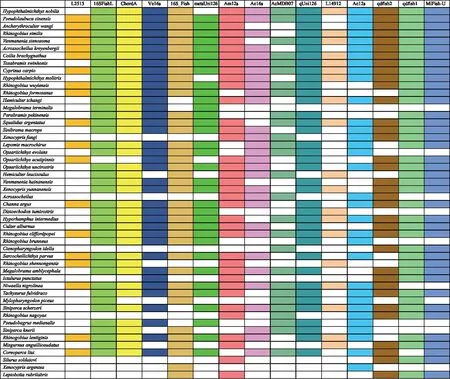
图6 不同引物监测到的鱼类物种差异图(丰度排名前50)Fig.6 Map of differences in fish species monitored by different primers (top 50 in abundance)
2.5 群落结构组成
基于不同引物所获得的高通量测序结果,绘制了不同物种的序列分布柱状图(图7)。引物L2513、Ve16s和qUni126扩增出的鱼类序列数只占总体序列数的50%左右,引物Ac12s扩增出的鱼类序列数占所有鱼类序列数的70%~80%之间,引物metaUni126、Am12s、qdfish2和qdfish1扩增出的鱼类序列数在80%~90%之间,如Am12s(84.42%);其余6对引物扩增的鱼类序列数占比都大于90%。在大部分引物所扩增出序列中,鱼类的序列都是最多的,但也有一部分引物的特异性不是很好,除了扩增出鱼类的DNA序列之外,也扩增出了一些其他类群物种的序列,如鸟类、爬行类、两栖类等。

图7 不同引物基于体外eDNA宏条形码分析所得的序列分布Fig.7 Sequence distribution of different primers based on in vitro eDNA metabarcoding analysis
将针对不同目的基因的宏条形码引物分为3类进行NMDS分析,结果如图8所示。NMDS分析计算出stress值为0.1,小于0.2,表明分析结果具有一定的可靠性,可以反映出不同引物监测到的鱼类物种间的差异程度。基于相同的eDNA样本,尽管不同的引物产生了不同的鱼类多样性特征,但不同组之间没有显著的差异。
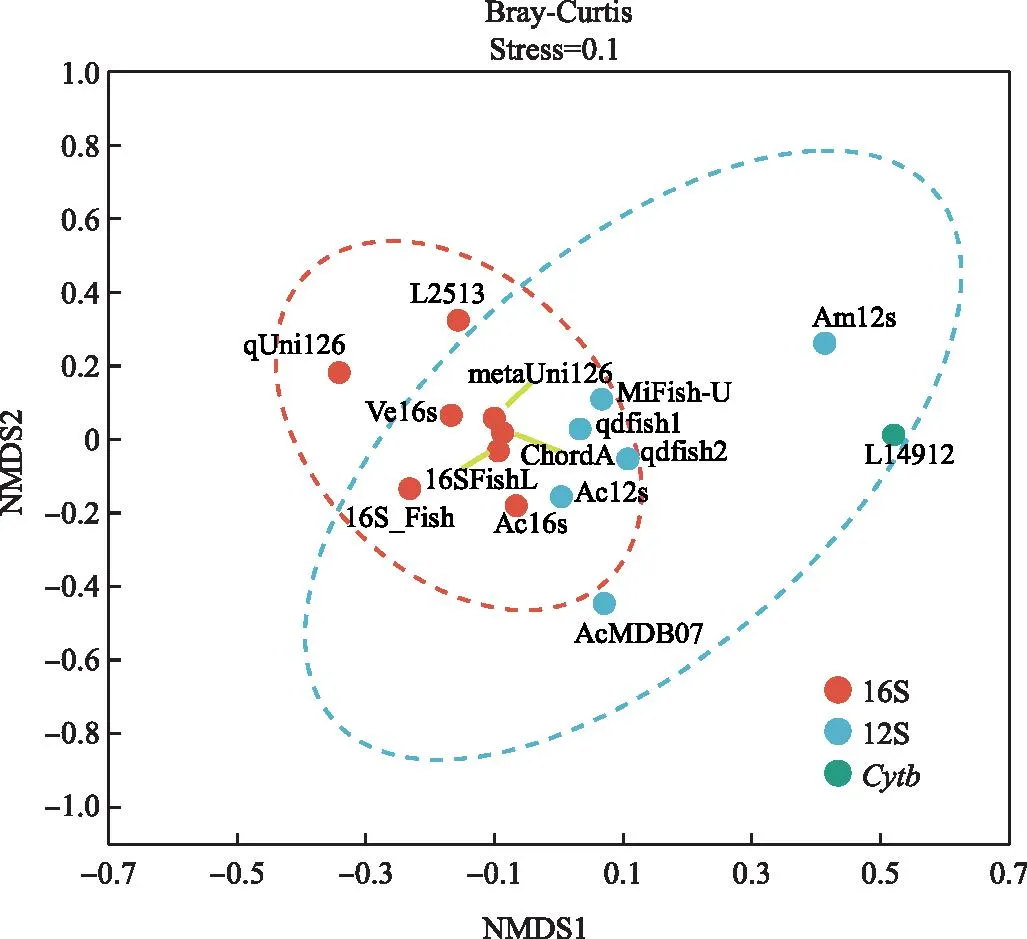
图8 鱼类群落结构的NMDS分析Fig.8 NMDS analysis of fish community structure
3 讨论
3.1 不同引物对于鱼类的覆盖度和分辨能力分析
对不同鱼类物种有着更广泛的覆盖范围并且能够有更大的区分程度是使用eDNA宏条形码对鱼类物种多样性进行监测的先决条件。通过计算机模拟PCR发现,不同的宏条形码引物对鱼类的扩增效率和分辨能力也有所不同。总的来说,COI和Cytb基因对于鱼类物种的分辨能力并不差,除了引物GFCC,其他引物扩增出的鱼类序列之间的遗传距离均处于平均值以上。此外,COI基因参考数据库所覆盖的物种也更加广泛[49]。然而,基于12S和16S基因设计的宏条形码引物比基于COI和Cytb的宏条形码引物所扩增出的鱼类物种数更多,因而能够区分出的鱼类物种数也更多。对此,以往的研究者也有着相似的发现,陈治等[50]通过对海南岛淡水鱼类的DNA条形码研究发现,对于鱼类物种判别能力最高的目标基因为COI,其次为16S和12S。但在实际的鱼类环境DNA研究中却很少用COI作为目标基因,其最大原因在于COI很难设计出DNA宏条形码通用引物[18],且也有研究表明,对水生动物DNA浓度低的水环境而言,靶向于鱼类COI基因的引物比12S表现较差[51],限制了COI引物在基于eDNA技术的鱼类多样性监测中的应用。
在模拟PCR后对其中的17对宏条形码引物作了进一步验证,分析结果显示,尽管17对引物在模拟PCR中的分析结果均显示良好,但在实际的扩增过程中,结果却各有不同。17对引物中有2对因未能够成功扩增出鱼类的序列而被删除,其余15对宏条形码引物最多的监测到了81种鱼类,最少的仅监测到了28种鱼类。由此可以看出,模拟PCR可以帮助我们增加对于引物本身的了解,但模拟PCR仍需要与实际的宏条形码分析结合起来。监测到鱼类物种数最多的5对引物中,有3对是基于16S rRNA设计的,有2对是基于12S rRNA设计的,这也表明了基于12S和16S基因都能够设计出较好的引物以用于eDNA研究。具体来看,不同引物对于鱼类的覆盖度和分辨能力也有所不同,如MiFish-U、16S_Fish、ChordA、16SFishL和qdfish1对鱼类的覆盖度更高一些,但从不同引物对扩增的鱼类物种的分辨能力来看,16SAR/BR、qUni126和Ve16s又是最好的。基于不同的宏条形码引物所获得测序结果,一方面不同的引物都监测到了一些相同的鱼类物种,使得它们之间可以相互印证,增加了研究的可靠性;另一方面,由于不同引物具有不同的特性和优缺点,互相补充能够最大程度地提高鱼类的检测概率,因此在今后的基于eDNA的物种多样性研究中,推荐采用多对宏条形码引物进行监测。
3.2 不同引物的特异性比较
对鱼类序列进行特异性扩增是鱼类环境DNA宏条形码引物选择的重点,如果对鱼类物种序列的特异性不够高,则在PCR扩增的过程中就可能过度扩增其他物种的序列,从而占用大量的测序通量并导致鱼类的测序量不足,无法获得足够的鱼类分类学信息[52]。本研究结果显示,几乎每一对引物都扩增出了非鱼类物种的序列,甚至有个别引物扩增出来的非鱼类物种的序列占比达到了所有物种的一半以上。Zhang等[16]的研究结果也发现,几乎所有鱼类宏条形码引物都扩增出了非鱼类物种的序列,这表明对于eDNA研究而言,寄希望于引物能够完全不扩增非目标物种似乎是不太现实的。通常而言,扩增出来的非目标物种大多是人类、鸟类、哺乳类、爬行动物、两栖动物和细菌等。由于人类活动、动物行为及水体流动等原因,eDNA中常常会涵盖非鱼类物种序列,因此在监测到鱼类物种的同时,监测到以上物种似乎不足为奇。近年来,一些研究者开始尝试使用阻断引物以解决这一问题[53]。然而,已有研究表明,使用阻断引物会降低eDNA宏条形码研究所能够监测到的目标物种的数量;据此,一些学者提出,若非目标物种的序列较低时,不建议使用阻断引物[16,54]。因此,为了避免非特异性扩增的出现并增加目标物种的监测概率:一方面需要充分了解目标分类群的DNA序列特征,选择适当的引物和扩增条件以最大程度地减少非目标序列的扩增;另一方面,对eDNA研究中难免会出现扩增出非目标物种这种情形,增加测序深度或许会是一种比较可行的解决办法。
3.3 不同引物扩增长度分析
由于eDNA数量少和易降解的特性,扩增片段长度较短的引物通常被认为可以提供更大的扩增成功率[55]。有研究者曾对水中的eDNA大小分布进行了研究,结果表明较短的线粒体DNA片段比长的DNA片段更为丰富,使用扩增子较短的引物可以获得更大的检测率[24,56]。但也有研究表明,较长的DNA片段能够提供更多有关序列的信息[57]。由于较少有研究通过使用实际环境样本系统地进行分析,往往也很难确定不同扩增子长度对于eDNA研究中物种多样性监测结果的影响。
过去有研究者曾对22对eDNA引物进行了综合的比较评价,结果显示Ac12S(约388 bp)和AcMDB07(约280 bp)效果最好,监测到的物种数最多,其次是MiFish-U(约171 bp)[16]。本研究中,用于实际验证的引物的扩增长度范围为100~500 bp,与以往研究者的研究结果有所不同,本研究中扩增片段长度更大的引物并没有获得理想中最好的效果,监测到物种数最多的反而是扩增片段长度在150~250 bp之间的引物,可能的原因是,在本研究中较短的DNA片段比长的片段数量更为丰富,从而获得了更大的扩增成功率,并监测到了更多的鱼类物种。
3.4 野外样品实验验证结果分析
本研究基于计算机模拟PCR筛选了17对宏条形码引物,并对千岛湖及其主要支流的eDNA样品进行分析后发现,引物MiFish-U、16S_Fish、ChordA、16SFishL、qdfish1监测到的鱼类物种数相对较多。与传统的鱼类资源调查结果相比,除了两种方法监测到的共有种之外,每一种引物都或多或少地监测到了传统方法未能捕获到的鱼类。这可能与以下两方面因素有关,一方面,栖息地间的连通及水文传输使采集到的eDNA样本中混入了来自其他水域的鱼类DNA[58-59];另一方面,参考数据库的不完善以及引物本身对于部分鱼类的分辨率较低,导致一些物种尽管被成功扩增,但未能够被鉴定出来[15]。
过去有研究评估了鱼类分布与eDNA之间的关系,结果表明eDNA通常提供了鱼类大尺度空间分布和生物量的“快照”[9]。其他一些研究也表明,基于eDNA推断的物种时空尺度分布格局会因河流和湖泊不同而异[13]。因此,eDNA在野外条件下的释放动态、降解和扩散模式有待进一步研究。
与传统方法相比,eDNA方法可以在不捕获目标物种的前提下进行生物多样性监测,极大地改变了动植物调查的方式[13]。而无论选择哪一种宏条形码引物,为实现生物的准确分类鉴定,可靠且完备的参考数据库是重中之重。公共数据库因为结合了数千项研究的成果,产生了大量的数据,而这些数据无法临时获得,因此当前的eDNA研究多依赖于与公共数据库(如GenBank或Mitofish)进行比对,从而实现物种的进一步确认。有研究表明,对于动物而言,GenBank中错误标记序列的比例非常低(在目水平上为0.05%,在属水平上<1%)[60],这说明它具备较高的可靠性,可以作为了解生物多样性的一大数据来源。然而,公共数据库中的序列多来自分散且目的不同的研究,也没有系统地补充和完善不同物种分类下DNA条形码的想法和动力。由此可知,尽管公共数据库中的序列信息仍然在不断增长,但分类学完整的条形码参考数据库的建设,尤其是有关本土鱼类物种序列的收集和整理仍然任重道远。
4 结论
本研究对29对宏条形码引物进行了筛选并在千岛湖进行了初步应用,从物种覆盖率和分辨率等角度评估了不同引物在基于eDNA的鱼类多样性研究中的适用性。本研究结果表明,eDNA技术灵敏度的确很高,可以极大地提高对于鱼类的监测效率。引物的好与不好一方面取决于其本身的设计(如上下游引物的长度、退火温度),另外一个方面也取决于数据库的完备性。因此,对引物进行先验性评估,然后再用于全面的生物多样性监测十分重要。由于不同引物对鱼类物种的覆盖范围和分辨能力也会有所不同,在这种情况下,通过使用多个引物来增加对于目标物种的覆盖度和检测概率,不失为一种比较好的解决方案。
猜你喜欢
英语世界(2023年10期)2023-11-17
海洋信息技术与应用(2022年1期)2022-06-05
少年文艺·开心阅读作文(2021年8期)2021-09-05
儿童时代·幸福宝宝(2020年9期)2020-09-08
小学科学(学生版)(2019年5期)2019-05-21
科学大众(中学)(2019年3期)2019-05-17
少儿美术(快乐历史地理)(2019年11期)2019-04-20
汽车观察(2018年10期)2018-11-06
小学生导刊(2017年13期)2017-06-15
金色少年(奇趣科普)(2016年8期)2016-09-21
