
基于面积归一化法对饲料中黄霉素总残留量的研究
2024-02-27 13:12卞华,曹莹,葛宇
中国粮油学报 2024年1期
卞 华, 曹 莹, 葛 宇
(上海市质量监督检验技术研究院1,上海 200233) (上海市兽药饲料检测所2,上海 201103)
黄霉素(Flavomycin)为磷酸化多糖类强极性抗生素, 主要由灰绿链霉菌厌氧发酵而成[1-3],只溶于水和甲醇等小分子溶剂中。黄霉素的抗菌作用机理是通过干扰细胞壁的结构物质肽聚糖的生物合成,从而抑制细菌的繁殖[4];黄霉素的促生长原理在于它能提高饲料中能量和蛋白质的消化,能使肠壁变薄,从而促进营养物质的吸收,有效提高饲料的利用率[5]。同时,黄霉素抗菌谱较窄, 主要对革兰氏阳性菌有效,不容易与别类抗生素产生交叉耐药性[6-8],曾一度被广泛用于畜禽和水产品饲料中。研究表明,长期服用黄霉素类药物对人的骨骼发育会有严重影响,甚至会产生造血功能障碍[9,10]。2005年欧盟农业部长会议[11]决定, 黄霉素等4种抗生素自2006年起禁止使用。日本肯定列表(Japan Positive List System)和美国环境保护局(EPA)等部门也相继出台了禁用规定。目前我国检验检疫部门已将黄霉素列为饲料和动物源性食品残留监控计划[12]。
黄霉素为多组分混合物,主要由5种活性成分黄霉素A、黄霉素A12、黄霉素C1、黄霉素C3、黄霉素C4组成,其化学特性和抗菌活性均十分相似。其中,黄霉素A为最突出的活性组分。然而黄霉素混合物的纯度不高,5种活性成分的分离成本极大,无法得到各自单独的标准品。迄今为止,黄霉素检测技术的发展并不理想,全球有关黄霉素检测的研究中均仅测定黄霉素A的含量,结果并不准确。我国《兽药质量标准》中也仅规定,可以通过检测黄霉素A的含量间接监控基质中黄霉素物质的添加量[13,14]。
黄霉素检测方法包括:微生物法[15,16]、高效液相色谱法[17,18]、液相色谱-串联质谱法[19]和超高效液相色谱-高分辨四级杆飞行时间质谱法[20]等。其中,微生物法属于筛选法,定量不准确,并且缺乏特异性;高效液相色谱法(HPLC)具有更好的特异性,在特定的波长下能够准确测定黄霉素的不同成分,但是其检出限较高,无法测定低含量的基质;液相色谱-串联质谱法(LC-MS/MS)具有高选择性、高精确度、高通量的独特优势,能精确测定基质中黄霉素A的含量,但因5类物质碎片离子的分子量相近,无法准确分离5种黄霉素活性成分,结果会有一定的误差;液相色谱-高分辨四级杆飞行时间质谱法(LC-Q-TOF-MS)可以对不同组分的精确分子量与特征碎片进行有效分离和定性分析,但无法准确定量。至今为止,各种检测方法都有一定的局限性。在全球范围中,均没有关于黄霉素权威性的标准,国际标准化组织(ISO)体系中也没有黄霉素相关标准的发布。
因此,本实验参考了国内外文献,根据不同类别饲料基质的特性,对前处理方法进行了研究,利用通透式固相萃取技术,结合高效液相色谱和液相色谱-串联质谱的特点,探索能同时满足黄霉素各活性成分均充分分离的方法,并准确测定黄霉素A的含量,经高分辨四极杆飞行时间质谱确认检测结果中黄霉素A的唯一性后,最终利用面积归一化法精确折算出基质中黄霉素的总残留量。该方法选择性强、灵敏度高,重复性好,可为饲料基质中黄霉素总含量检测标准的制定提供参考。
1 材料与方法
1.1 实验材料与仪器
黄霉素固体标准品(11015-37-5, 92%)。折算后,将黄霉素标准储备液质量浓度配制为1 000.0 μg/mL,黄霉素标准中间液质量浓度配制为100.0μg/mL(现配现用)。甲醇、乙腈(色谱纯);甲酸、氨水(色谱纯);乙酸铵(分析纯);甲酸铵(分析纯)。Oasis PRiME HLB 6cc/200 mg。
e2695 Separations Module液相系统,2998 PDA detector检测器,XEVO TQ-XS三重四极杆串联质谱仪,电喷雾离子源(ESI),AcquityR UPLC H-CLASS 液相色谱系统,SYNAPT XS High Resolution Mass Spectrometer四级杆,Synapt HDMS质谱分析系统,MSZO 204S型电子天平,JM-03D-40超声波清洗机,D-91126多管涡旋振荡器,Centrifuge 5804高速离心机,Preekem-N1全自动氮吹浓缩仪,Milli-Q超纯水系统。
1.2 试样处理
提取:称取试样5 g (准确至0.01 g),置于50 mL离心管中,准确加入25 mL体积分数0.1%甲酸甲醇溶液,涡旋振荡5 min,超声10 min,在10 000 r/min离心机上,离心5 min,取上清液置于50 mL容量瓶中,重复步骤,合并上清液,用体积分数0.1%甲酸甲醇溶液定容至刻度。
净化:吸取10 mL提取液至Oasis PRiME HLB通透式固相萃取小柱中,减压过滤并保持流速为1~2滴/s,直接收集过滤液。取1 mL净化后的过滤液经0.22 μm有机相滤膜过滤后供液相色谱-串联质谱分析。
1.3 标准溶液配制
1.3.1 面积归一法(供高效液相色谱分离)
将黄霉素标准中间溶液(100 μg/mL)用甲醇稀释成质量浓度为50 μg/mL的标准工作液,用于测定标样中各活性成分的峰面积。
1.3.2 基质加标标准工作曲线(供液相色谱-串联质谱定量)
将黄霉素标准中间溶液100 μg/mL用甲醇逐级配制成质量浓度为1.0、5.0、10.0、25.0、50.0、100.0μg/mL的标准系列溶液,称取与试样基质相应的阴性样品5.0 g,分别加入各标准系列溶液0.5 mL,与试样同时提取和净化。
1.4 色谱条件
1.4.1 高效液相色谱
色谱柱:DiKMA Platisil 5 μm ODS, 250.0 mm×4.6 mm;柱温:35 ℃;进样体积:20 μL;流动相体积比:20 mmol/L乙酸铵溶液(甲酸调节pH 至5.0)∶乙腈=55∶45;紫外检测器:258 nm。
1.4.2 液相色谱-串联质谱
液相色谱条件:色谱柱: Atlantis T3(100.0 mm×2.1 mm, 3 μm);柱温:35 ℃;进样体积:10 μL。流动相A:5 mmol/L 乙酸铵溶液(pH 5.0);流动相B:乙腈;流速0.4 mL/min。梯度洗脱程序:0.0~1.0 min,10%B;1.0~1.1 min,10%B~95%B;1.1~4.0 min,95%B;4.0~4.1 min,95%B~10%B,4.1~7.0 min,10%B,质谱仪器条件:电喷雾负离子模式(ESI-),多反应监测(MRM);毛细管压力:3.0 kV;椎孔电压:40 V;离子源温度:150 ℃;脱溶剂气温度:550.0 ℃;脱溶剂气流速:1 000 L/h;反吹气流速:150 L/h;碰撞气流速:0.15 mL/min;雾化气压力:0.7 MPa。母离子、子离子、去簇电压(DP)、碰撞气能量(CE),见表1。

表1 黄霉素A化合物的母离子、子离子、去簇电压和碰撞能
2 结果与分析
2.1 高效液相色谱条件的优化
2.1.1 色谱柱的选择
黄霉素为强极性弱酸性混合物,在反相柱上不易保留。普通改良的C18色谱柱并不适用于黄霉素混合物。黄霉素C1、黄霉素C3、黄霉素C4几乎没有得到有效的分离,并且,黄霉素A12与黄霉素A被同时洗脱,形成很强的前伸效应,导致峰形很宽。结果表明,黄霉素混合物能在更适合酸性盐流动相体系下的铂金柱DIKMA Platisil 5 μm ODS, 250.0 mm×4.6 mm中得到较好的分离,提供更好的峰型(图1),其可能与铂金柱中去活化的酸性游离硅羟基固定相对5种活性组分在四氢吡喃基团上不同的R1、R2、R3基团有不同的分子间作用力(表2),从而产生了不同的分离结果。
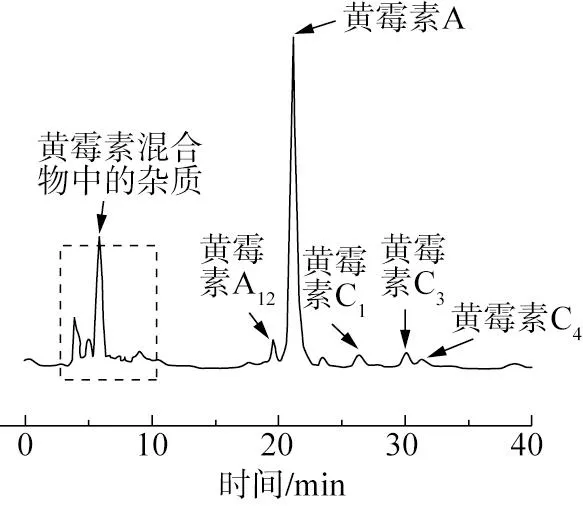
图1 黄霉素混合物的高效液相色谱分离图

表2 黄霉素活性成分的结构信息
2.1.2 流动相的确定
以DB32/T 1279—2008[21]标准为依据,将20 mmol/L甲酸铵和乙酸铵分别作为预选水相,乙腈和甲醇作为预选有机相。实验表明,甲酸铵和乙酸铵均属于弱酸弱碱盐,阴离子和阳离子均水解,水溶液显中性,无法分离得到目标物,需要将溶液环境用甲酸调整到pH 5.0后能得到较明显的目标峰。通常,在pH 3.5~5.5时,相同浓度的乙酸铵-酸溶液比甲酸铵-酸溶液缓冲能力略强,最终选择20 mmol/L乙酸铵为水相。同时,当有机相采用甲醇时,目标物的保留时间都更早,导致黄霉素A12与黄霉素A,黄霉素C3与黄霉素C4都包裹在一起,乙腈的极性相对较弱,更适合于黄霉素混合物的分离。
另外,当有机相的比例大于48%时,黄霉素A12与黄霉素A分离效果理想,但黄霉素C1、黄霉素C3、黄霉素C4的出峰时间明显前移,与主峰黄霉素A同时被洗脱;当有机相的比例小于43%时,黄霉素A12会延后,同样会与黄霉素A同时被洗脱。最终,当有机相的比例为45%时,能得到最清楚和最稳定的分离结果。5个化合物在6次平行性实验得出保留时间的偏差在0.16 s以内。
2.1.3 黄霉素标准品各组分含量的确定
根据国内外饲料生产企业所提供的信息,在黄霉素被广泛使用时,主要由4家公司制备合成黄霉素类抗生素。利用高效液相色谱条件测定黄霉素标样(50 μg/mL),分离后的黄霉素各活性组分经面积归一化法和统计模型加权平均后(表3),得出黄霉素A质量分数为86.2%,RSD为0.66%。值得一提的是,黄霉素杂质峰在258 nm处的光谱图均与黄霉素主要活性成分的光谱图不符(图1),已在配制标准储备液时进行了折算,不在研究考虑范围之内,不纳入面积归一法中进行计算。

表3 黄霉素A在不同黄霉素标准品中的质量分数(n=6)
2.2 液相色谱串联质谱条件的优化
2.2.1 色谱条件的优化
黄霉素极性较强,普通的C18柱子容易受到基质效应的影响,峰形不理想。Atlantis®T3色谱柱使用高纯度硅胶及双键键合C18技术,并对填料的孔径大小、端基封口以及 C18的键合密度进行优化,从而使色谱柱具有对黄霉素A保留能力强,对低pH条件下的色谱十分有利,能保证黄霉素A具有良好的分离稳定性,色谱柱峰形尖锐对称,灵敏度更高,保留时间也更适合。
研究分别选取乙腈、甲醇作为备选有机相,以超纯水、体积分数0.1%氨水为备选水相,流动相两两配比。结果表明甲醇和乙腈作为有机相,都能较好地分离出黄霉素A,但乙腈相对于甲醇,系统整体的柱压下降了13%,目标峰的保留时间也更为稳定。在负离子模式下,微量的氨水可以提供ESI源在Q1四极杆内离子化所需的负离子,从而提高离子化效率,得到更好的色谱峰形和更高的灵敏度[22]。但本实验发现,在高pH值的流动相环境下,黄霉素A的响应值反而降低,主要与黄霉素呈弱酸性有关。相反,实验同样尝试了pH 5.0的5 mmol/L乙酸铵溶液作为水相,其使得黄霉素A的离子峰形更加尖锐,响应也更高。
黄霉素是以黄霉素A(C69H108N5O34P)、黄霉素A12(C68H106N5O34P)、黄霉素C1(C62H96N5O28P)、黄霉素C3(C63H98N5O28P)、黄霉素C4(C63H98N5O29P)为主的多组分混合物。考虑到5种活性成分的分子质量均在1 000 u以上,将配制的质量浓度为1.0 μg/mL的黄霉素标准溶液通过注射泵注入质谱仪中进行一级质谱全扫描(分子量扫描范围:50~2 000 u),黄霉素在电离过程中产生2个明显的母离子,分别为[M-2H]2-m/z1 580.7和[M-2H]2-m/z789.9,其中[M-2H]2-m/z789.9的响应远高于[M-2H]2-m/z1 580.7(图2),因此在质谱优化过程中以[M-2H]2-m/z789.9作为黄霉素的母离子。根据文献可知,[M-2H]2-m/z789.9为黄霉素A的母离子,因此实验利用液相色谱—串联质谱主要测定黄霉素混合物中黄霉素A的含量。
同样地,将母离子[M-2H]2-m/z789.9进行二级质谱全扫描(分子质量扫描范围:50~800 u),只找到了2对离子[M-2H]2-m/z789.9/553.9和[M-2H]2-m/z789.9/575.9,且2个通道几乎都没有杂质干扰(图2)。因此实验对2对离子进行CE优化,并选取相对丰度较高且稳定的特征碎片离子[M-2H]2-m/z789.9/575.9作为定量离子,[M-2H]2-m/z789.9/553.9作为定性离子对,以满足欧盟657/2002/CE指令对禁用药物定性检测的规定,即满足1个母离子和至少2个子离子共4个识别点数的要求[23]。图3为黄霉素A的多反应监测色谱图。

图2 黄霉素标样的一级扫描和二级全扫描质谱图

图3 黄霉素A的TIC和MRM色谱图(100.0 ng/mL)
另外,为了验证黄霉素A的色谱分离具有唯一性,不受其他组分的影响。实验还利用LC-Q-TOF-MS高分辨四极杆飞行时间质谱仪在相同的流动相条件下研究了黄霉素混合物的分离结果。最终发现,黄霉素A在9.087min保留,此时无其他黄霉素活性成分(图4)。由上而下依次为8.933 min处黄霉素A12[M-2H]2-m/z1 567.6/782.8的母离子全扫图;9.087 min处黄霉素A [M-2H]2-m/z1 581.6/789.9的母离子全扫图;9.606 min处黄霉素C1[M-2H]2-m/z1 389.6/693.8的母离子全扫图;9.995 min处黄霉素C3[M-2H]2-m/z1 403.6/700.8的母离子全扫图;10.279 min处黄霉素C4[M-2H]2-m/z1 419.6/708.8的母离子全扫图。LC-Q-TOF-MS能较好地分离5种活性成分,但定量的稳定性却不是很理想,相对标准偏差在16%以上。

图4 黄霉素混合物在超高分辨质谱仪中分离的结果(100.0 ng/mL)
2.2.2 提取条件的选择
通过对黄霉素A分子结构的研究,可知其仅溶于水和低分子醇。实验考察了甲醇和甲醇/水对饲料基质加标样品回收率的影响(禽浓缩饲料、禽配合精饲料、禽添加剂预混合饲料、畜浓缩饲料、畜配合精饲料、畜添加剂预混合饲料)。结果表明,当提取溶剂为甲醇,6种基质提取效率均较高,接近80%,而甲醇/水提取效率较低(见表4)。可能是因为黄霉素为弱酸性物质,水为弱电离介质,水中的羟基对黄霉素A的作用力要远小于甲醇中的羟基。实验进一步考查了体积分数10%氨化甲醇溶液和0.1%甲酸甲醇溶液对于黄霉素A回收率的影响。0.1%甲酸甲醇溶液有助于提升黄霉素A的回收率,甲酸除了能降低提取液的pH值,还具有协助甲醇沉淀蛋白类杂质的作用,可以进一步降低基质效应。另外,考虑到黄霉素A在电离过程中会失去电子,本实验尝试用10%氨化甲醇溶液作为提取液,但回收率有一定的下降,这是因为氨化甲醇作为提取液更适用于肉制品基质中黄霉素A的提取[24,25],对于基质更为复杂的饲料基质来说,氨水提取时,会造成饲料黏糊成小颗粒,降低提取回收率的同时,影响结果的稳定性。因此,实验最终选择10%甲酸甲醇溶液作为提取剂。
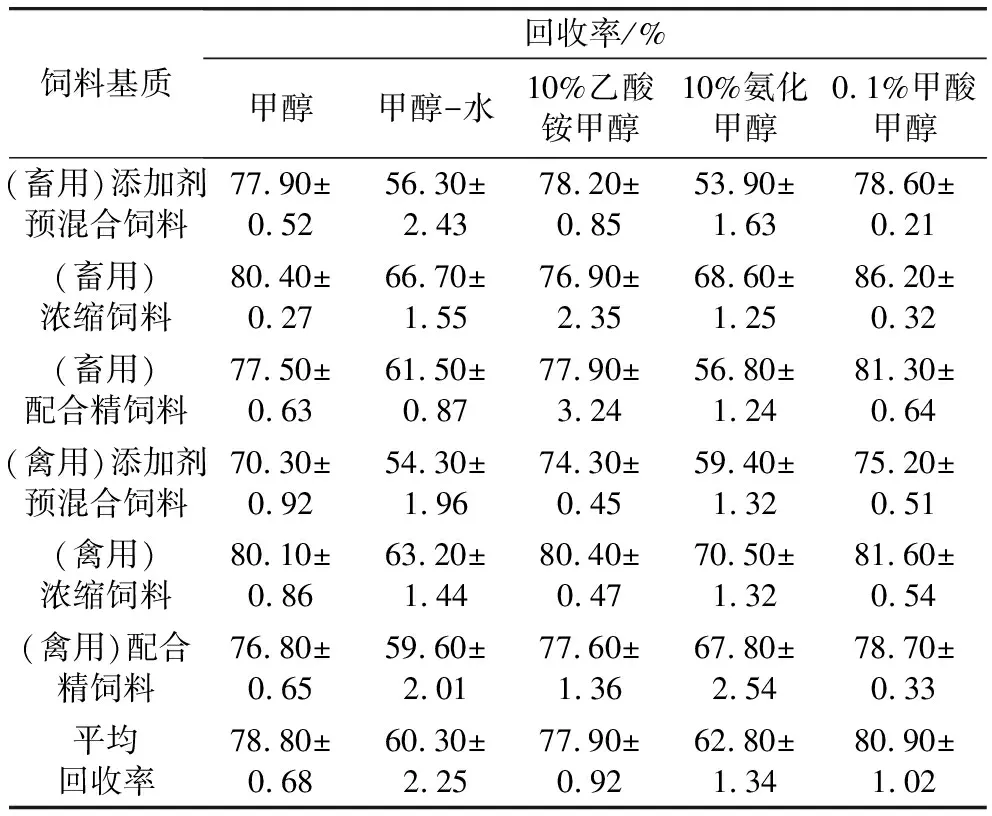
表4 不同提取剂对6种饲料基质中黄霉素A提取回收率的影响(n=6)
2.2.3 净化条件的确定
基质经过0.1%甲酸甲醇提取后,吸取10 mL上清液,选择C18、Oasis HLB, Oasis PRiME HLB和氨基Amino-NH2固相萃取小柱进行净化条件考察(饲料基质中,抗生素的添加含量通常较高,无需浓缩)。由表5可见,只有Oasis PRiME HLB固相萃取对于黄霉素A的回收率有一定的提升作用。
利用普通C18和Oasis HLB净化提取液,得到的黄霉素A回收率均有一定的下降,无法充分地去除杂质。主要由于黄霉素A用甲醇提取,而常用的SPE(Solid-Phase Extraction)通常用强极性溶剂(如甲醇)洗脱,导致目标物大多在上样时没有保留。另外,在洗脱过程中,因为饲料基质的复杂性,导致SPE柱经常堵塞,对结果的稳定性也有一定的影响。而Oasis PRiME HLB能有效地将杂质保留在填料中,其通过筛板、填料以及制作工艺上的特殊设计,使样品经提取后直接过柱净化、收集,将黄霉素A在净化过程中的损失降低到了最低。6种基质的总体回收率上升了1.6%,特别能将添加剂预混合饲料基质的回收率提升至80%左右。同时,整个净化过程较为稳定,样品更加洁净,基质效应更小;Amino-NH2适合于极性化合物,弱阴离子交换萃取,但对于黄霉素A的保留结果也不理想,并且伴有较强的基质效应。因此,本实验选用Oasis PRiME HLB固相萃取作为净化方式。
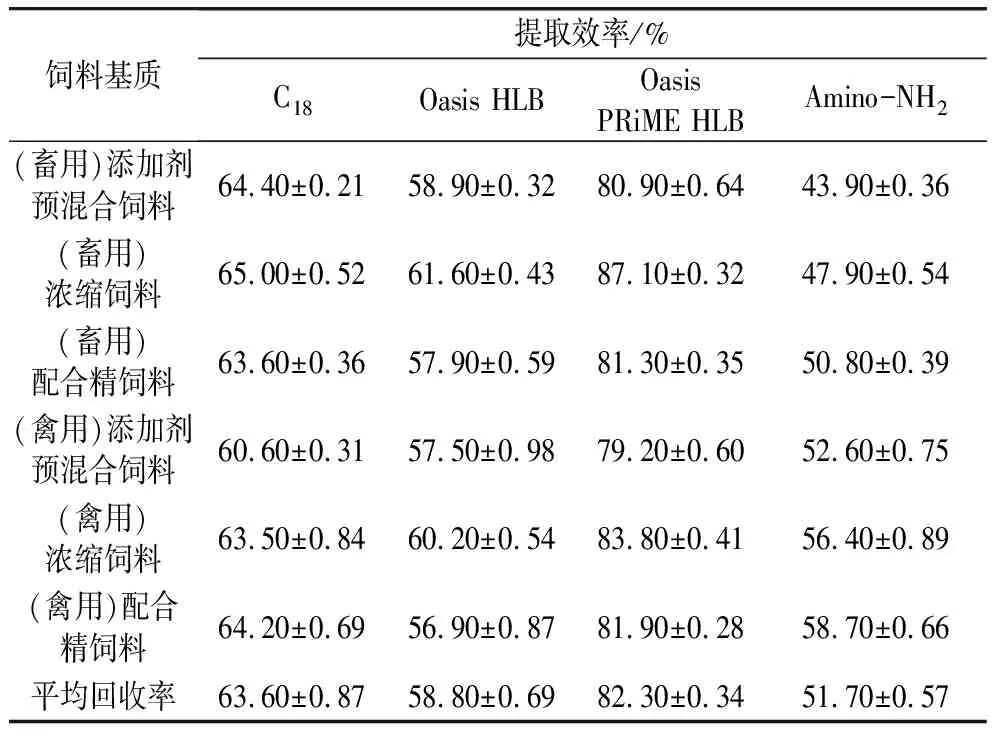
表5 不同净化条件对饲料中黄霉素A的提取效率的影响(n=6)
2.3 方法学评价
2.3.1 检出限、定量限和线性关系
饲料基质复杂,本实验选择畜用(牛)和禽用(鸡)各3类基质的空白样品作为考察对象。在空白基质中添加一定浓度的黄霉素对照品,按照该方法进行测定。在液相色谱-串联质谱仪上,依据定量离子信号与噪声比例,即s/n≥3的含量为检出限(LOD),s/n≥10的含量为定量限(LOQ)的原则,确定质谱方法的检出限为100 μg/kg,定量限为250 μg/kg。黄霉素A在10.0~1 000.0 ng/mL范围内线性良好。其中,畜浓缩饲料基质的线性相关系数(R2)为0.999 8,线性方程为Y=246.899X+1 164.82。图5为(牛)预混饲料基质的空白和加标总离子流图。
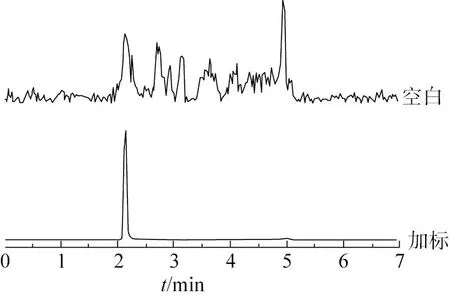
注:加标水平:5 LOQ。图5 (畜)预混料基质色谱图
2.3.2 回收率和精密度
选取畜和禽的空白基质进行为期1个月的加标回收率及精密度实验,样品分别添加低、中、高3个浓度的标准溶液(6基质×6平行×4批次)。不同饲料基质中黄霉素A的平均加标回收率为69.69%~88.60%,RSD为0.96%~3.38%。
由表6可知,浓缩饲料基质的回收率结果较为理想,回收率较高,稳定性也较强。浓缩饲料的颗粒较大,比表面积较小,目标物更容易被有机溶剂提取[26];基质相对复杂的添加剂预混合饲料为一种或多种微量组分与稀释剂或载体按要求配比,均匀混合后制成的中间型配合饲料产品。添加剂预混合饲料颗粒最细,目标物通常较难被提取,基质效应导致禽类和畜类基质整体的回收率均偏低,约3%;配合精饲料主要由能量饲料和蛋白质饲料组成,基质本身最为复杂,其中的颗粒大小不同,但平均比表面积较小[27]。因此,配合精饲料与添加剂预混合饲料的平均回收率没有显著差异,但配合精饲料结果的相对标准偏差更小,结果更为稳定。实验结果均满足食品质量规范技术要求,因此可以说明本方法的准确性和精密度均较为理想,能够满足黄霉素精确定量的检测要求。
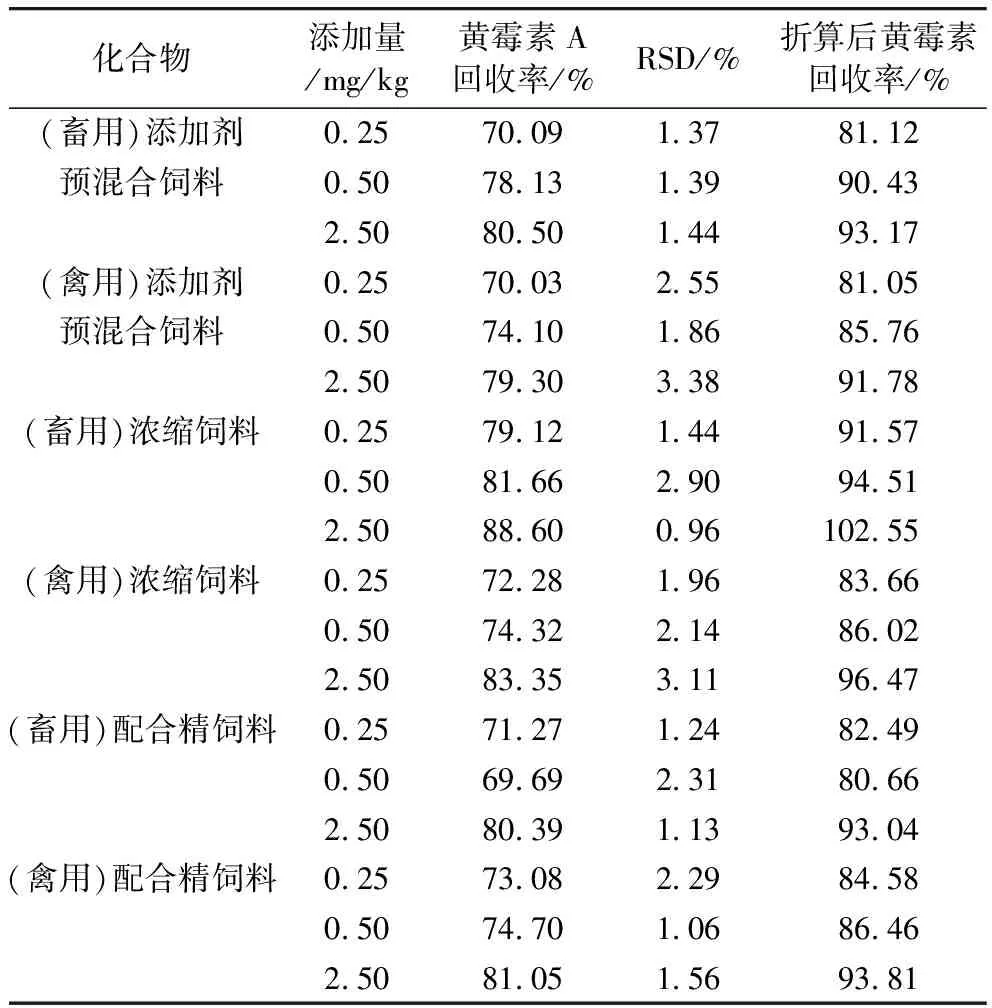
表6 不同饲料基质中黄霉素A及其折算后黄霉素的加标回收率及相对标准偏差(n=6)
3 结论
黄霉素A在液相色谱-串联质谱法测定下的线性范围为10.0~1 000.0 ng/mL,基质曲线线性相关性均良好,线性相关系数均>0.998 6。各饲料基质的最低检出限为100 μg/kg,定量限为250 μg/kg,加标回收率在69.69%~88.60%之间,RSD在0.96%~3.38%之间。同时,黄霉素5种活性成分可以通过高效液相色谱法分离而得,经面积归一化法可知黄霉素A的所占比例为86.2%,RSD为0.66%,实验结果稳定,可以用黄霉素A来折算黄霉素的含量,最终得出各类饲料基质中,黄霉素类抗生素残留量的回收率在80.66%~102.55%之间。结果符合《兽药残留检测标准操作规程》和GB/T 23182—2008相关标准要求。因此,该方法适用于常用饲料中黄霉素的测定,对黄霉素总残留量检测在饲料领域标准的制定具有较高的参考价值。
致谢
感谢农业农村部农产品质量安全监管司和上海市食品质量安全检测与评价专业技术平台对本实验的支持,感谢农业部委员会专家对本实验方法的认可和优化意见,并感谢上海市兽药饲料检测所的专家和美国普渡大学食品科学与工程学院的教授对本实验的指导和建议。
猜你喜欢
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
世界最新医学信息文摘(2021年12期)2021-06-09
四川蚕业(2020年4期)2020-02-10
中国蜂业(2018年4期)2018-05-09
当代化工研究(2016年6期)2016-03-20
中外医疗(2015年16期)2016-01-04
河北工业科技(2015年4期)2015-02-27
食品工业科技(2014年9期)2014-03-11
无机化学学报(2014年3期)2014-02-28
