
基于LC-MS/MS 技术的水蛭配方颗粒中7 种碱基和核苷成分分析方法的建立
2024-03-01 10:45程春雷
环境化学 2024年1期
姜 俊 程春雷 杨 昊
(山东省食品药品检验研究院(国家药品监督管理局仿制药研究与评价重点实验室),济南,250000)
水蛭为水蛭科动物蚂蝗(WhitmaniapigraWhitman)、水蛭(HirudonipponicaWhitman)或柳叶蚂蝗(WhitmaniaAcranulataWhitman)的干燥全体,具有破血通经、逐瘀消癥等功效,临床上具有很高的药用价值[1].针对水蛭药材,采用液相色谱法对其进行黄嘌呤、尿苷、尿嘧啶、次黄嘌呤中1—4 种碱基及核苷成分的含量测定已有相关文献报道[2−4].碱基是嘌呤和嘧啶的衍生物,是合成核苷、核苷酸和核酸的基本组成单位.核苷是是核酸合成的前体,具有保护神经、抗病毒和抗菌等生物活性.李桃运用多种色谱分离手段从水蛭中提取并分离鉴定出38 个化合物[5],荆文光等从水蛭甲醇提取物中分离鉴定了18 个化合物[6],除上述4 种成分外,还包含其他几种成分,如腺苷、腺嘌呤、次黄嘌呤苷(肌苷),但未均建立此类成分的含量测定方法.
水蛭配方颗粒是由水蛭饮片经水加热提取、浓缩、干燥、制粒而成的一种颗粒.目前,水蛭配方颗粒暂未出台相应的国家药品标准,广东、山东、湖南等地公布了水蛭配方颗粒的省级质量标准,这些标准中含量测定项主要涉及抗凝血活性、水蛭胺C、次黄嘌呤的测定.除此之外,未见水蛭配方颗粒中多种碱基和核苷成分同时测定的相关报道.碱基和核苷化合物绝大部分是强极性的小分子,仅依靠传统的疏水作用机制难以进行较好的色谱保留,该类化合物也一直是分析技术的难点.本试验通过多重保留机制的液相色谱-质谱联用技术,建立同时快速测定水蛭配方颗粒中7 种碱基和核苷类成分的含量方法,该方法具有分析快速、准确、灵敏度高的特点,可为水蛭配方颗粒甚至水蛭药材的质量评价提供依据.
1 实验部分
1.1 试剂与仪器
试剂:尿嘧啶、次黄嘌呤、腺苷、腺嘌呤、肌苷对照品(上海源叶生物科技有限公司),黄嘌呤对照品(中国食品药品检定研究院),尿苷对照品(上海安谱实验科技有限公司),纯度均大于98%.水蛭配方颗粒来源于广东一方制药有限公司,每袋1.5 g,每1 g 配方颗粒相当于2 g 药材.
仪器:超高效液相色谱仪Nexera X2 与三重四极杆质谱LCMS-8050 联用系统(岛津,日本).
1.2 样品的制备
混合标准溶液的制备:精密称取7 种碱基和核苷对照品各适量,除黄嘌呤用20 mmol·L−1氢氧化钠溶解外,其余均采用10%甲醇溶液溶解,最终用10%甲醇溶液配制成系列浓度的混合标准溶液,待上机分析.
样品前处理:取水蛭配方颗粒5 袋,研细,称取约0.1g(精确到0.0001g),加入10%的甲醇溶液100 mL,涡旋混匀1 min,50 ℃超声30 min,4000 r·min−1离心10 min,上清液用0.22 μm 的尼龙滤膜滤过,取续滤液即得.样品的稀释:精密量取制备好的样品续滤液1 mL,用10%甲醇溶液定量稀释至10 mL.
1.3 实验条件
液相色谱条件:色谱柱为Discovery®HS F5-3(150 mm×2.1 mm,3 μm),流动相:A 为0.15%甲酸水溶液,B 为甲醇;流速0.35 mL·min−1,梯度洗脱,0—1min,2%—5%B,1—5 min,5%B—50%B,5—5.5 min,50%B—90%B,5.5—7 min,90%B,7.01—10 min,2%B;柱温40 ℃,进样体积:1 μL.
质谱条件:ESI 正负离子模式同时监测;接口电压:+0.5 kV(正离子),−0.5 KV(负离子);雾化气流速:氮气3.0 L·min−1;加热气流速:空气15 L·min−1;干燥气流速:氮气5 L·min−1;脱溶剂管温度:150 °C;加热模块温度:300 °C;离子源接口温度:350 °C;扫描模式:多反应监测(MRM);驻留时间:25 ms;MRM 参数见表1.
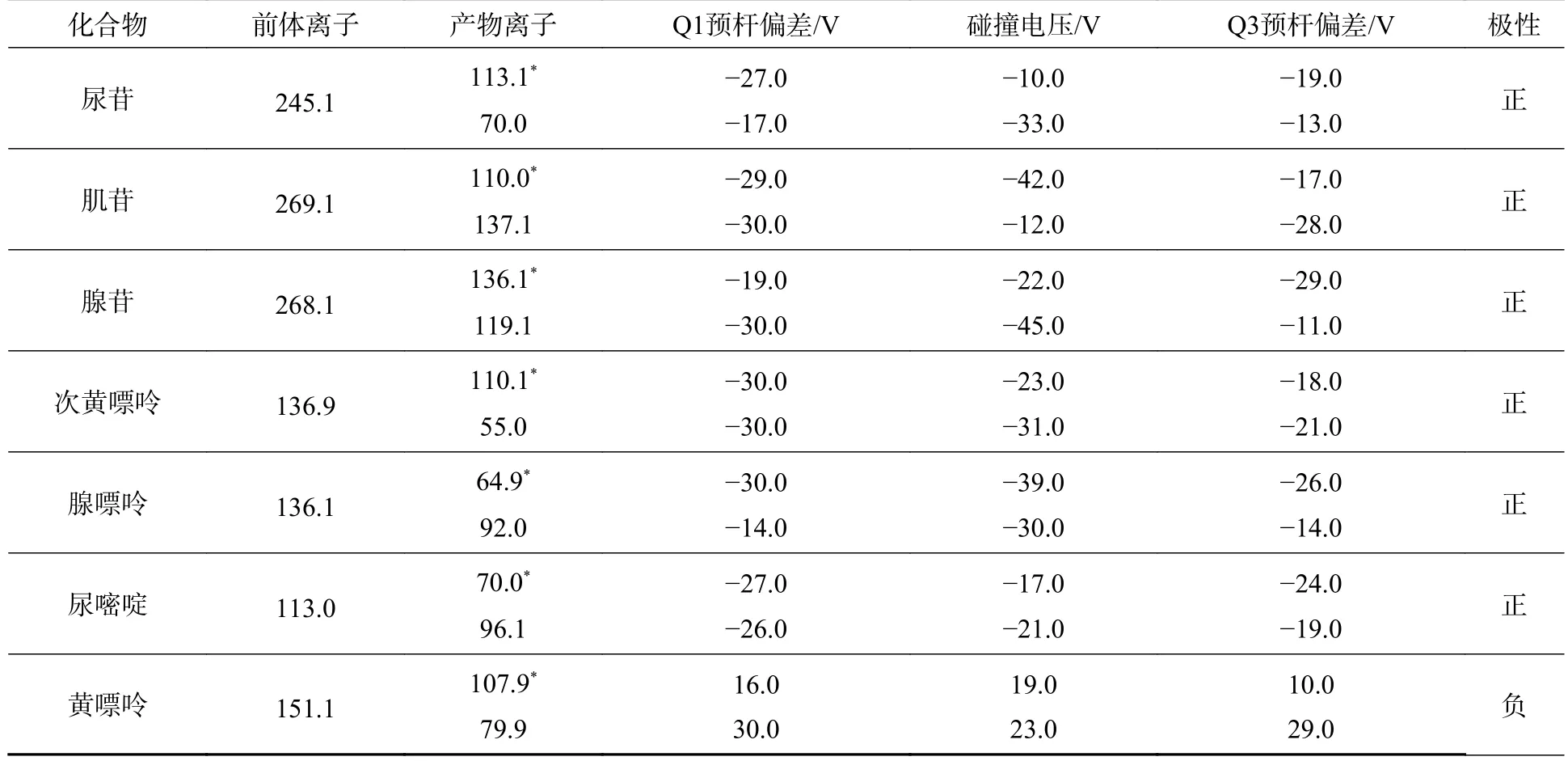
表1 7 种碱基和核苷的MRM 参数列表Table 1 MRM parameters for 7 nucleobases and nucleosides
2 结果与讨论
2.1 样品前处理条件的选择
依据待测化合物的溶解性及参考相关文献[4,7],本实验尝试了使用水、10%甲醇、70%甲醇、0.1%甲酸及10 mmol·L−1氢氧化钠在50 ℃下进行超声提取30 min,考察不同溶剂的提取效率.结果显示:70%甲醇、0.1%甲酸及10 mmol·L−1氢氧化钠提取条件下,肌苷的峰面积偏低,且70%甲醇提取条件下色谱峰会展宽.水和10%甲醇对大部分化合物的提取效率相当,但10%甲醇对腺苷的提取效率要优于水.综合考虑,确定样品溶液的制备方式为10%甲醇50 ℃条件下超声30 min.
2.2 分析条件的优化
本实验曾比较了不同流动相系统对色谱峰及质谱响应的影响,如乙腈-0.02%甲酸和1 mmol·L−1乙酸铵水溶液、乙腈-0.1%甲酸水溶液、甲醇-0.1%甲酸水溶液、甲醇-0.15%甲酸水溶液.最后确定采用甲醇-0.15%甲酸水溶液的流动相体系,该体系下各化合物色谱保留较好且质谱信号较强.
7 种待测碱基和核苷化合物的属于极性较强的化合物,传统反相色谱保留效果欠佳.董萌等尝试用亲水色谱对食品中的碱基、核苷和核苷酸类物质进行色谱保留[8].亲水色谱对强极性化合物保留较好,但要得到较好的峰形和重复性,方法开发的难度往往比普通反相色谱的难度要大.本实验对比了Discovery®HS F5-3、Shim-pack GIST C18 和ShimNex UP C18 色谱柱对分析测试的影响.结果显示Discovery®HS F5-3 色谱柱对待测化合物的保留最好,且质谱响应信号相对更高.Discovery®HS F5-3 是一款在硅胶基质上键合了五氟苯基的色谱柱,提供了疏水作用、氢键作用、离子作用等多重保留机制,对强极性化合物的保留和异构体的分离优于常规C18 柱.
2.3 精密度试验
将10 ng·mL−1的混合对照品溶液连续进样6次,尿苷、肌苷、腺苷、次黄嘌呤、腺嘌呤、尿嘧啶和黄嘌呤7 种化合物测得峰面积的RSD 分别为4.50%、5.83%、1.84%、3.35%、4.27%、2.08%、3.54%,表明精密度良好.
2.4 线性范围和灵敏度
取系列浓度的混合标准溶液上机测定,以对照品浓度为横坐标(X)、峰面积为纵坐标(Y)绘制校准曲线,得到7 种碱基和核苷的线性方程、判定系数.以各化合物信噪比(S/N)约为3时计算对应的浓度为检出限(LOD),各对照品信噪比(S/N)约为10 时对应的浓度为定量限(LOQ),结果如表2 所示.由表2 可知,7 种碱基和核苷在相应的浓度范围内具有良好的线性关系,判定系数(R2)≥0.996,检出限在0.13—1.18 μg·g−1范围内,定量限在0.43—3.94 μg·g−1范围内,该方法灵敏度较高.
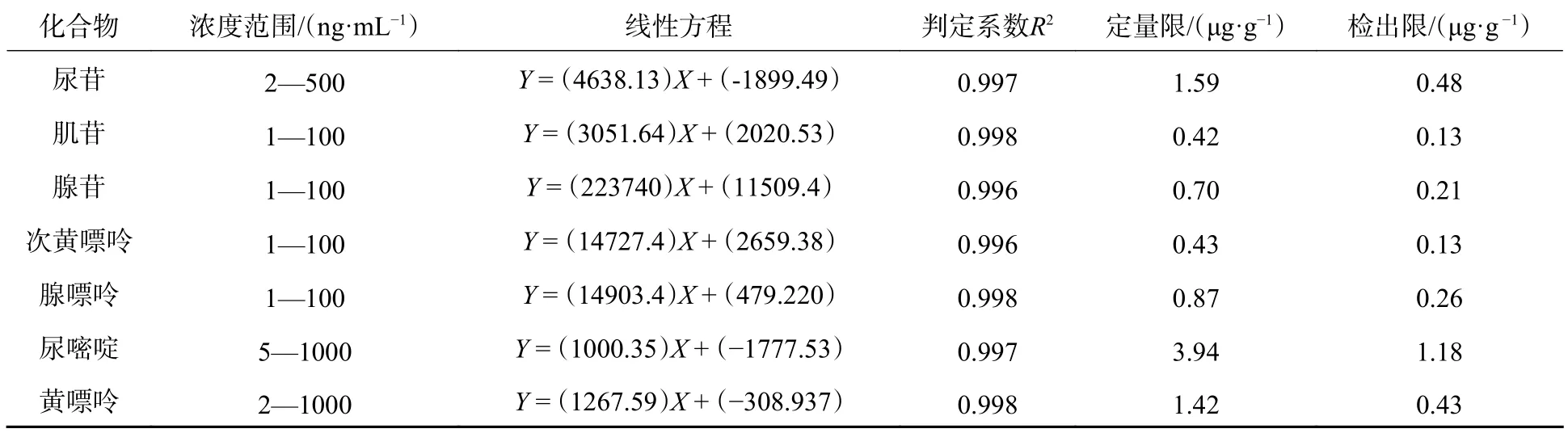
表2 7种碱基和核苷的线性范围及灵敏度Table 2 Linear range and sensitivity of 7 nucleobases and nucleosides
2.5 加样回收率试验
取“1.3”项下样品溶液上机测定,以外标法计算7 种碱基和核苷的含量.尿苷、肌苷、腺苷、次黄嘌呤、腺嘌呤、尿嘧啶和黄嘌呤的含量分别为41.42、7.11、1.24、314.87、4.89、55.83、53.95 μg·g−1.本品中尿苷、次黄嘌呤、尿嘧啶和黄嘌呤的含量相对较高,肌苷、腺苷和腺嘌呤有检出,但含量较低.另精密称取已测知含量的水蛭配方颗粒约0.1g,精密加入适量的对照品储备液,按“1.3”项方法制成加样回收样品溶液,平行制备6 份.进行次黄嘌呤回收率测试时,将以上加样回收样品溶液稀释10 倍上机进行测定,外标法计算平均回收率,结果见表3.7 种碱基和核苷的平均回收率在87.09%—105.81%之间,RSD%在1.35%—4.70%之间,表明方法的准确性高.
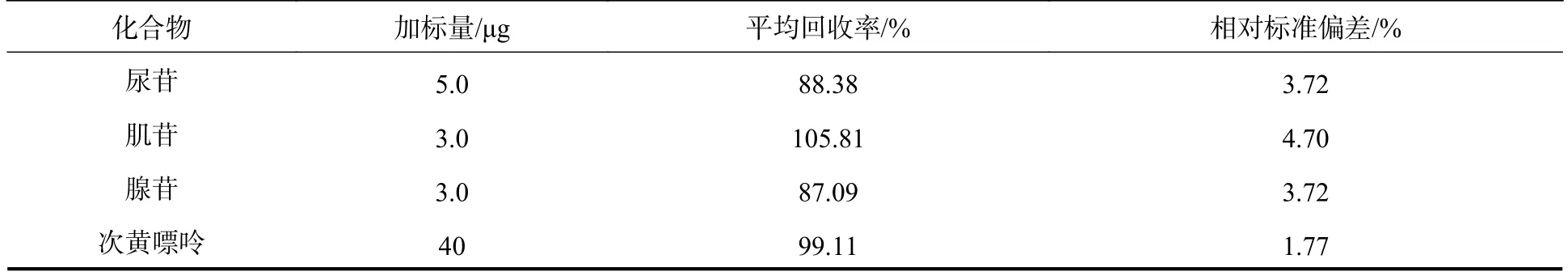
表3 7种碱基和核苷的加样回收率(n=6)Table 3 The recovery data of 7 nucleobases and nucleosides

续表3
2.6 残留实验
高浓度混合标准溶液(1000 ng·mL−1)分析完成后,进样10%甲醇试剂空白,考察残留情况.结果表明,7 种碱基和核苷化合物检测通道中均无明显目标化合物干扰.
3 结论
本文建立了一种使用岛津超高效液相色谱仪Nexera X2 和三重四极杆质谱仪LCMS-8050 联用测定水蛭配方颗粒中7 种碱基和核苷成分的分析方法.该方法分析速度快,重复性好,灵敏度和准确度较高,适合水蛭配方颗粒中碱基和核苷类化合物的快速定量检测,可为水蛭配方颗粒的质量评价提供参考.
猜你喜欢
现代食品(2022年3期)2022-03-24
基层中医药(2021年10期)2021-06-05
基层中医药(2020年12期)2020-07-22
中成药(2018年10期)2018-10-26
作文评点报·小学五、六年级(2018年28期)2018-05-18
中成药(2017年5期)2017-06-13
中成药(2017年5期)2017-06-13
山东农业科学(2017年2期)2017-03-15
河南科技学院学报(自然科学版)(2016年3期)2016-03-30
中国当代医药(2015年9期)2015-03-01
