
模仿IgG4 相关淋巴结病的多中心型Castleman 病:易误诊病例的鉴别诊断及文献复习
2024-03-10 13:42李春艳刘国荣常丽君李秀博丁文双
广州医药 2024年2期
李春艳 刘国荣 常丽君 李秀博 丁文双
1 广州市第一人民医院,华南理工大学第二附属医院病理科(广东广州 510180)
2 南方医科大学附属茂名医院病理科(广东茂名 525000)
Castleman病(Castleman’ disease,CD)是一种罕见的淋巴增生性疾病,根据临床表现分为单中心型(unicentric Castleman’ disease,UCD)和多中心型(multicentric Castleman’s disease,MCD)[1]。MCD根据是否感染卡波西肉瘤相关疱疹病毒/人类疱疹病毒8(Kaposi’s sarcoma-associated herpesvirus,KSHV/human herpesvirus-8,HHV8)分为KSHV/HHV8感染相关性、特发性和POEMS相关MCD(POEMS-MCD)。特发性多中心型Castleman病(idiopathic MCD,iMCD)分为浆细胞型(plasma cell type-iMCD,PC-iMCD)及高血管型(hyaline-vascular-iMCD,HV-iMCD),其中以PC-iMCD多见,PC-iMCD的淋巴结表现为淋巴滤泡增生、生发中心萎缩,滤泡间区成熟浆细胞片状增生[2-3]。IgG4相关淋巴结病有多种生长模式,其组织形态不具有特异性,其中I型生长模式与PC-iMCD有较多重叠之处,PC-iMCD可伴有大量IgG4阳性浆细胞浸润且血清学IgG4水平异常升高,当PC-iMCD形态及实验室检查同时符合IgG4相关淋巴结病诊断标准时,两者的鉴别非常困难,但两类疾病的治疗方案及预后完全不同,必须将其区分[4-5],这给病理诊断带来很大挑战,容易造成误诊。本文旨在分享1例与IgG4相关性淋巴结病的组织病理学相似的PC-iMCD,结合其临床症状、影像学特征及实验室检查并回顾相关文献以探讨2种疾病的鉴别要点,旨在加强临床医师对两种疾病的认识,避免误诊、误治。
1 资料与方法
1.1 病例资料
患者为48岁女性,因口干、多饮18年,皮肤瘙痒2年,加重1个月入院。既往有贫血、2型糖尿病病史。体格检查:未见明显异常。实验室检查显示多项体液免疫指标升高:IgA 13.4 g/L;IgG 91 g/L;IgM 2.57 g/L;IgE >6 000 ng/mL;补体C3 1.2 g/L;补体C4 0.239 g/L;Kappa轻链2 230.0 mg/dL;Lambda轻链1 290.0 mg/dL;血清IgG4水平14.7 g/L。胸部CT平扫及多平面重建:双侧腋窝、纵隔、上腹部腹膜后多发肿大淋巴结,考虑血液系统疾病可能;脾脏多发占位,性质待定。PETCT:全身多发淋巴结代谢稍活跃,全身多处骨髓弥漫性代谢稍活跃,考虑淋巴瘤多发浸润,建议结合病理检查;脾大伴有稍高密度结节,代谢不高,疑浸润。
1.2 苏木素-伊红(HE)染色
4%中性甲醛液固定标本,石蜡包埋切片(厚度=3 μm)经脱蜡、水化后用全自动HE染片机(赛默飞,型号:Shandon Varistain Gemini)行HE染色。
1.3 免疫组织化学(免疫组化)染色及EB病毒原位杂交
采用4%中性甲醛液固定标本,石蜡包埋切片(厚度=3 μm)常规脱蜡水化后,采用Leica全自动免疫组化染色机(设备型号:BenchMark ULTRA型)进行免疫组织化学染色。本研究标记抗体包括CD3、CD5、CD20、CD79a、CD10、CD21、CD56、CD38、CD138、CD43、Bcl-2(B cell lymphoma protein-2)、Bcl-6、MUM-1[Multiple myeloma(MM)antigen 1)]、S-100、IgG4、IgG、Ki-67,上述抗体均为即用型抗体,购自广州安必平医药科技股份有限公司,均设置阳性对照。EB病毒原位杂交试剂盒购于北京中杉金桥生物技术有限公司,根据试剂盒步骤进操作,同时设置阳性对照。
2 结 果
2.1 镜下HE形态
淋巴滤泡增生,生发中心萎缩(图1A),部分套区淋巴细胞呈“洋葱皮”样围绕生发中心排列(图1B),滤泡间区扩张伴血管增生,其内见大量成熟的浆细胞呈片状浸润,灶区见含铁血黄素沉积(图1C-D),浆细胞及淋巴细胞均未见明显异型性。
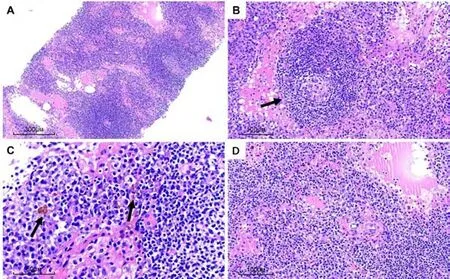
图1 PC-iMCD 镜下组织形态
2.2 免疫表型
滤泡树突网CD21阳性,显示生发中心萎缩(图2A);滤泡间区成熟浆细胞CD79α、CD38、CD138(图2B)、MUM1及CD43阳性;滤泡间区IgG4阳性浆细胞>100/高倍视野,IgG4阳性细胞数/ IgG阳性细胞数比值>40%(图2C-D);Kappa及Lambda轻链显示非限制性表达(图2E、F);生发中心B淋巴细胞CD20、CD79α、CD10及Bcl-6阳性,Ki-67增殖指数20%;T淋巴细胞CD3、CD5阳性。
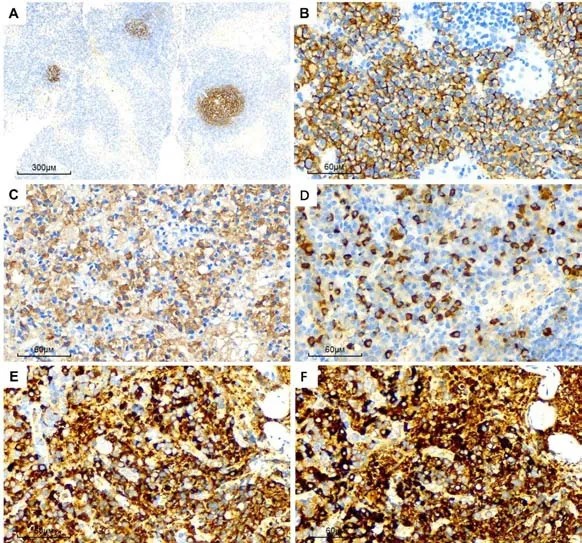
图2 PC-iMCD 免疫表型
2.3 EB病毒检测
EB病毒编码RNA原位杂交阴性(图3A)。

图3 PC-iMCD 原位杂交
3 讨 论
本研究报道了1例少见PC-iMCD的淋巴结病变,其组织学特征与IgG4相关淋巴结病变-MCD样亚型组织学改变类似,镜下均表现为淋巴滤泡增生,滤泡间见大量成熟浆细胞浸润。同时该患者淋巴结内IgG4计数>100/高倍视野、IgG4/IgG比例>40%、血清IgG4水平14.7 g/L(IgG4相关疾病临界值1.35 g/L),其形态学、免疫组化及实验室检查均符合IgG4相关淋巴结病变诊断标准。两种疾病的治疗措施及生存预后有较大差异,在此情况下,两类疾病的的鉴别极为重要,结合文献复习所总结的鉴别要点见表1。
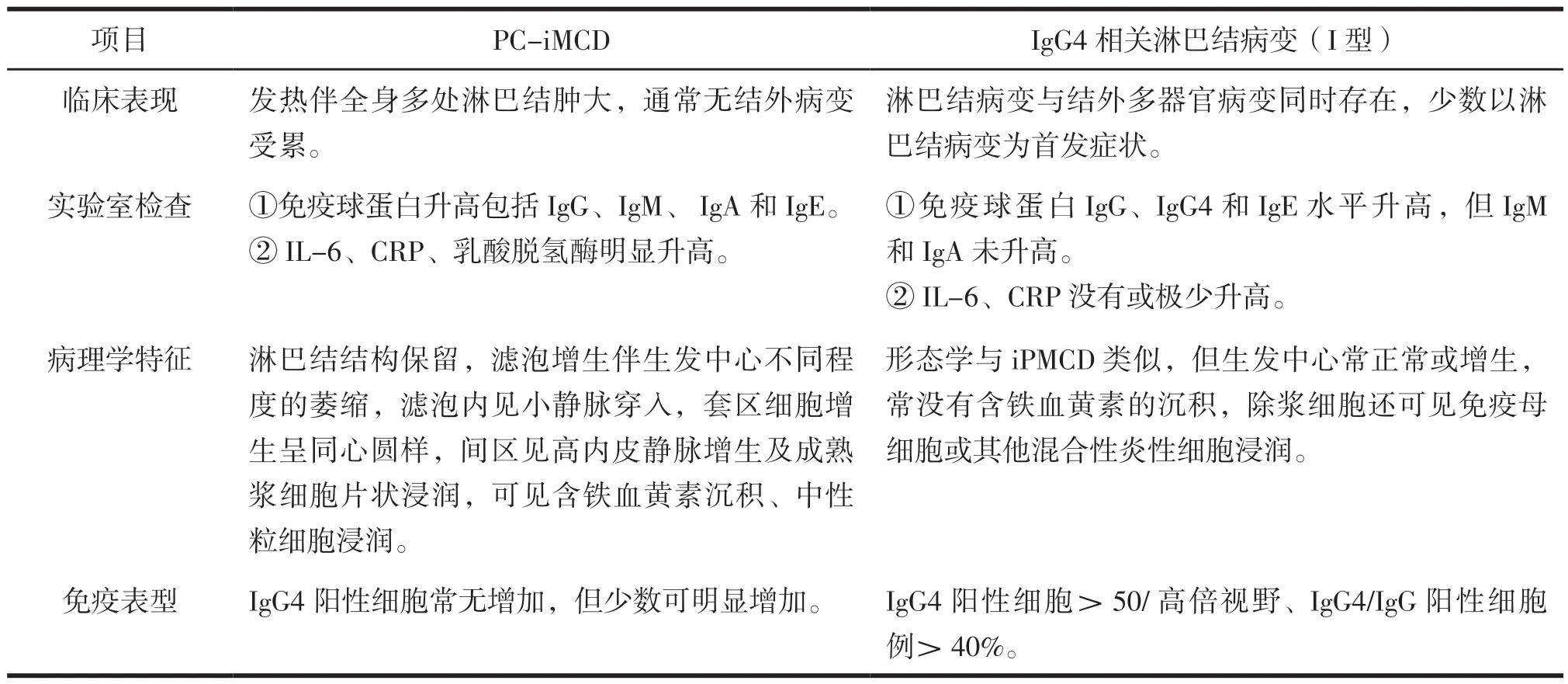
表1 PC-iMCD与IgG4相关淋巴结病变(I型)的鉴别总结
IgG4相关病变(IgG4-related disease,IgG4-RD)为系统性疾病,临床表现全身多处器官受累,最常累及胰腺、唾液腺、泪腺、腮腺和肺[3]。IgG4-RD确诊依靠综合性诊断,修订版(2020版)诊断标准[6]中包括临床影像、血清学和病理诊断标准,其中特征性的病理改变包括:①受累组织内大量淋巴浆细胞浸润,伴纤维化;②IgG4+浆细胞/IgG+浆细胞比值>40%,且IgG4+浆细胞>10/高倍视野(诊断淋巴结病变需>50/高倍视野[7]);③席纹状纤维化和闭塞性静脉炎,其中IgG4+浆细胞绝对值需与器官特异性IgG4-RD 诊断标准中临界值相结合。80%的IgG4-RD伴全身多处淋巴结肿大,大部分IgG4相关淋巴结病变与结外病变同时存在,仅少数病例以淋巴结病变为首发症状,随后出现典型结外组织学改变[8]。目前IgG4相关的淋巴结病变分为5种组织学类型,多中心Castleman病样(Ⅰ型)、滤泡增生(Ⅱ型)、滤泡间扩张(Ⅲ型)、生发中心进行性转化(Ⅳ型)和炎性假瘤样(Ⅴ型)[4,9]。IgG4相关淋巴结病变组织学不具特异性,如I型淋巴结改变与MCD相似,镜下均表现为淋巴结结构保留,髓窦扩张开放,滤泡增生伴生发中心不同程度的萎缩退变,滤泡内见小静脉穿入,滤泡间区可见高内皮静脉增生及成熟浆细胞大量浸润[4]。值得注意的是,IgG4+浆细胞浸润并非IgG4相关淋巴结病变特有,部分自身免疫性疾病相关淋巴结病变,MCD等也可出现IgG4+浆细胞大量增生,少数情况下可伴血清中IgG4水平升高,因此诊断IgG4相关淋巴结病变需与伴有类似组织学和血清学改变的疾病进行鉴别[6]。
C D 是一种罕见的淋巴增殖性疾病,依据临床表现分为单中心型(U C D)和多中心型(MCD)。不伴KSHV/HHV8感染的MCD称之为iMCD,iMCD包括PC-iMCD和HV-iMCD。PCiMCD发病与IL-6异常升高有关,患者表现为发热伴全身多处淋巴结肿大、血清蛋白异常升高、高乳酸脱氢酶等,镜下改变主要为淋巴滤泡增生伴生发中心萎缩,部分生发中心可见小静脉插入,髓窦扩张,滤泡间区大量成熟浆细胞片状浸润[10]。部分PC-iMCD患者淋巴结病变可伴有IgG4+浆细胞浸润及血清IgG4水平升高,韩国一项纳入87例CD的研究发现,11.5%(10/87)病例可满足IgG4相关淋巴结病变诊断标准[11]。Sato等[12-13]分析总结IgG4相关淋巴结病变与MCD组织学鉴别要点如下:①MCD患者生发中心常萎缩,IgG4相关淋巴结病变则正常或增生;②MCD滤泡间区成熟浆细胞为片状增生模式,IgG4相关淋巴结病变则细胞成分混杂,表现为成熟浆细胞与浆样细胞及免疫母细胞的增生;③IgG4相关淋巴结病变内可见嗜酸性粒细胞浸润,MCD未见。
为了更加精准的诊断IgG4相关病变,日本学者Sato等[14]提出了IgG4-RD的排除性诊断标准,目的在于排除PC-iMCD等具有相似组织学改变的疾病。该排除性诊断标准临床特征包括:①血清CRP持续升高(≥10 g/L);②血清IgA升高;③血清IgM升高。病理特征包括:①浆细胞片状增生;②铁血黄素沉积;③中性粒细胞浸润。随后,Nishikori等[3]通过纳入58例PC-iMCD的资料进一步验证此排除标准,发现其中20.5%(8/39)淋巴结病变和42.1%(8/19)肺病变可符合IgG4-RD诊断标准,通过排除性诊断标准,16例患者均可排除IgG4-RD,该排除性诊断标准的灵敏度和特异度分别为100%和93.1%。结合该排除性诊断标准,本文报道的病例可排除IgG4相关淋巴结病变,符合PC-iMCD的诊断标准。
Xia等[15]把组织学改变同时满足IgG4-RD和MCD诊断标准的这类少见病变创新给予IgG4-CD暂定名称,作者从临床表现、实验室检查及临床治疗预后角度进行了回顾性分析。IgG4-CD病例少见,其发病率约占IgG4-RD的2.5%(15/534),IgG4-CD发病部位兼具MCD和IgG4-RD的特征。本组数据中全部IgG4-CD病例(15/15)累及多处淋巴结,同时87%病例(13/15)伴淋巴结外多部位累及,常见于舌下腺、腮腺、胰腺及副鼻窦等部位。与MCD相比,IgG4-CD血红蛋白、嗜酸性粒细胞数量、血清IgG4水平及IgG4/IgG比例偏高,而CRP、IL-6水平等偏低。IgG4-CD患者可接受单纯糖皮质激素治疗或联合糖皮质激素/免疫抑制剂治疗,该类患者治疗效果及预后同IgG4-RD较一致,生存预后明显优于MCD。该作者首次创新性地从临床及治疗角度提出IgG4-CD疾病名称,为解决这类难以鉴别的疾病提供了新的诊断和治疗思路,但该组数据的病理学资料描述不够详尽,同时受样本量少的限制,该暂定名称的疾病在病理学及风湿免疫学领域的接受度可能受到挑战。
伴血清IgG4升高的PC-iMCD与IgG4相关淋巴结病(Ⅰ型)组织形态、免疫表型特征及血清学改变均有重叠,单纯依靠形态学难以将两者进行鉴别,容易造成误诊、误治。两者鉴别需结合IgG4相关病变诊断标准、排除性诊断标准、临床表现及相关实验室检查综合判断。
猜你喜欢
影像研究与医学应用(2021年3期)2021-11-30
中国临床医学影像杂志(2021年5期)2021-08-13
天津医科大学学报(2021年3期)2021-07-21
肝博士(2021年1期)2021-03-29
实用临床医学(2018年9期)2018-12-04
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
成都中医药大学学报(教育科学版)(2016年1期)2016-01-22
实用皮肤病学杂志(2015年4期)2015-12-22
天津医科大学学报(2015年2期)2015-12-22
