
CsPbX3钙钛矿纳米晶中Mn(Ⅱ)离子的可控掺杂及其发光性能研究
2024-03-11 12:14陈洁訚哲王立瑾李宇王帅冰林欧阳唐爱伟滕枫
发光学报 2024年2期
陈洁, 訚哲, 王立瑾, 李宇, 王帅冰, 林欧阳, 唐爱伟, 滕枫
(北京交通大学 物理科学与工程学院, 北京 100044)
1 引言
全无机卤化钙钛矿最早是由Loredana Protesescu等于2015年采用高温热注射法合成[1],由于其简单的合成工艺、高光致发光量子产率(PLQY)、较窄的半峰宽以及可调谐的PL光谱成为当前最具潜力的光电材料之一[2-5],相比于制备成本较高的硒化镉(CdSe)[6]和磷化铟(InP)[7-8]等纳米晶表现出独特的优势。过去的十年中,CsPbX3(X= Cl,Br,I)全无机钙钛矿蓬勃发展,不论在制备方面,还是在发光二极管、光电探测以及太阳能电池等应用领域均取得了巨大进展,具有非常广阔的前景[9-13]。然而,其稳定性和Pb毒性问题极大地限制了该材料的实际应用[14-16]。因此,科研人员致力于寻找无毒或低毒的化学元素以替代传统钙钛矿中有毒的Pb元素。目前最常用的替代元素包括Sn、Ge、Bi和Sb等[17-19],通过将Pb全部取代,合成了多种具有相似结构的无铅钙钛矿材料。然而,Pb的高离子电导率是钙钛矿纳米晶具有高离子性的关键因素[20],有助于Pb基钙钛矿保持高PLQY和稳定性[21];此外,由于CsPbX3钙钛矿具有较小的Stokes位移,其发射谱比较单调,存在较强的重吸收效应[1]。因此,使用其他金属离子部分替代Pb是一个可取途径。
近几年来,使用过渡金属离子部分取代CsPbX3钙钛矿中的Pb多有报道[22-24]。其中,使用Mn2+进行部分掺杂的方法得到了广泛关注。Mn2+的掺杂既可以减少钙钛矿中的Pb含量,降低毒性,又可以提高钙钛矿材料的稳定性以及光致发光效率,还能够引入新的激子跃迁途径从而改变材料的发射特性[25-28]。虽然在实验过程中原料的投入量是一定的,但实际参与反应的量、Mn2+的掺杂比例根据实验条件而定,不易控制。因此,探索纳米晶的制备过程实现Mn2+的可控掺杂具有重要意义。此外,掺杂结构与合金结构在组成与发光方式方面均存在明显不同,但目前尚未有研究在CsPbCl3∶Mn2+体系中的掺杂与合金化过程进行明确区分[29-31]。本文通过一步和两步热注射法合成了不同Pb∶Mn投料比的CsPbCl3∶Mn2+纳米晶(NCs),对Mn2+的实际掺杂含量进行了大范围和精确调控,有效控制了Mn2+的掺杂速率和实际掺杂含量,同时将所得样品纳米晶明确区分为合金结构和掺杂结构。合金结构中引入离子较多,且由多种物质组成;而掺杂结构只是微量引入掺杂离子,只会稍改变其晶格参数,而不会改变其原有的晶体结构。两种结构中Mn2+的引入均显著提高了材料的稳定性和光致发光量子产率,由3%分别提高至77%和66%。同时对两种方法调控Mn2+掺杂量的反应机制进行了探究。为了实现光谱的可调性,使用一步热注射法合成了一系列不同Br-含量的CsPb-(Clx/Br1-x)3∶Mn2+,将纳米晶发光颜色从蓝色调至橙色。
2 实验
2.1 油酸铯前驱体的合成
CsPbCl3∶Mn2+钙钛矿均采用热注射法合成,根据原料MnCl2加入的时间不同,将其分为一步法和两步法。将2.5 mmol碳酸铯(Cs2CO3)、2.5 mL油酸(OA)与40 mL十八碳烯(ODE)混合置于四口烧瓶中,温度升至120 ℃,真空环境下加热30 min,之后通N2,保持120 ℃加热,直至完全溶解。使用前需120 ℃预热。
2.2 一步法
将0.7515 mmol氯化铅(PbCl2)、0.7515 mmol氯化锰(MnCl2)和20 mL ODE、2 mL OA、2 mL油胺(OM)混合置于四口烧瓶中,在真空环境下升温至120 ℃,并保持1 h;随后将温度升至180 ℃,快速注入1.6 mL油酸铯,立即冰水浴。将所得液体在6000 r/min的速度下离心15 min,倒掉上清液,将所得沉淀用5 mL正己烷溶解,再离心一次,将上清液倒入西林瓶中进行保存。
2.3 两步法
首先使用上述一步法合成纯CsPbCl3样品。然后准备MnCl2的前驱体溶液:将MnCl2(0.7515 mmol)与7.5 mL ODE、1.5 mL OA、1.5 mL OM混合,真空情况下120 ℃加热至完全溶解,使用前120 ℃预热。最后将CsPbCl3加热至120 ℃并保持5 min,向其中快速注入MnCl2的前驱体溶液,立即冰水浴;随后6000 r/min离心15 min,倒掉上清液,使用6 mL正己烷溶解沉淀,再离心一次,将上清液放在西林瓶中进行保存。调整MnCl2的使用量,得到不同Pb∶Mn的样品。也可先合成CsMn-Cl3后加入PbCl2。
2.4 CsPb(Clx/Br3-x)∶Mn2+的合成
基于一步法,固定反应物Pb2+与Mn2+的量均为0.7515 mmol,调整卤素比例(Cl∶Br),使得Br-取代Cl-的含量分别为25%、50%、75%、100%。
2.5 样品表征
材料的形貌和尺寸使用JEM-1400(100 kV)透射电子显微镜(TEM)和JME-F200(200 kV)高分辨率透射电子显微镜(HRTEM)进行表征。材料的X射线衍射(XRD)谱使用辐射源为Cu Kα(λ= 0.154056 nm)的Bruker D8 Advance X射线衍射仪测定。样品的X射线光电子能谱(XPS)使用辐射源为300 W、Al Kα的VG ESCALAB 220i-XL能谱仪进行表征。样品的元素含量采用Nex-ION300x电感耦合等离子体质谱(ICP-MS)测定。材料的吸收光谱通过Ocean Optics USB 2000 分光光度计进行测量。材料的荧光寿命使用FLS1000瞬态稳态荧光光谱仪测量。材料的发光光谱和相对光致发光量子产率(PLQY)均采用FLUORAT-02-PANORAMA荧光光谱仪测定。以罗丹明B为标准参比物(QY为97%,溶剂为乙醇)测定PLQY,计算公式如下:
其中,η为量子产率;I为PL强度测量的积分面积;n为折射率(正己烷为1.375,乙醇为1.362);A为激发波长处的吸光度,通常小于0.10。下标R表示罗丹明B, S表示所得样品,所有数据均在365 nm激发波长下测量。所有测量均在室温下进行。
3 结果与讨论
3.1 CsPbCl3∶Mn2+ NCs的组分调控及发光性能
研究发现,Pb2+与Mn2+的投料比对于CsPbCl3∶Mn2+纳米晶的合成起着决定性作用。因此,在本工作中,保持油酸铯的注入量一致,首先采用一步热注射法合成了不同Pb∶Mn的纳米晶。图1(a)为所制备的CsPbCl3∶Mn2+钙钛矿纳米晶的TEM和HRTEM图像,随着Mn2+投料量的增加,纳米晶的尺寸明显减小。一方面是由于半径较小的Mn2+已经成功取代Pb2+,导致晶格收缩;另一方面是由于Mn2+引入后,表面电荷密度增加,由于电荷斥力的影响,带负电荷的Cl-向表面扩散的过程会被抑制,从而导致尺寸减小[32]。HRTEM图像显示清晰的晶格条纹,表明所得纳米晶具有良好的结晶性。Pb∶Mn=1∶1.0时的晶格间距(d=0.39 nm)略小于Pb∶Mn=1∶0时的晶格间距(d=0.40 nm)[21],更加表明了Mn2+的引入。为了验证引入Mn所处的状态,我们对其进行了结构和成分分析。图1(b)显示了所得纳米晶的XRD图像,与标准卡片(PDF#180036)相比,含有Mn2+的CsPb-Cl3与不含Mn2+的CsPbCl3均具有相同的四方结构。我们可以明显观察到,随着Mn含量的增加,衍射峰向大角度移动,这表明半径较小的Mn2+取代了NCs中半径较大的Pb2+,导致晶格收缩。这也进一步证明了Mn2+的成功引入。此外,随着Mn2+投料量的增加,出现了CsMnCl3的衍射峰(红色虚线框标注),两种物质相互混合,这说明采用一步法合成的纳米晶为合金结构。利用XPS对样品中各元素的表面组成和化学状态进行了分析(如图1(c)、(d)所示)。不同Pb∶Mn投料比所合成的CsPbCl3∶Mn2+NCs都出现了额外的Mn信号,并且Mn和Pb均以二价形式存在。随着Mn2+含量的增加,Pb2+的信号向高结合能轻微红移,这主要是由于Mn2+取代Pb2+后晶场环境发生了改变。以上结果有力地证明了Mn2+进入晶格,占据了Pb2+的位置。
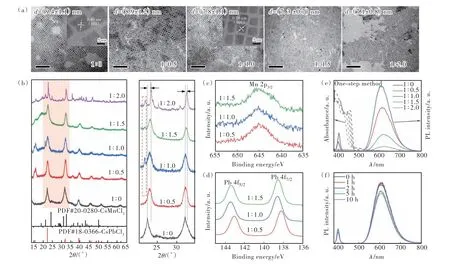
图1 PbCl2定量时,一步法所合成的不同Pb∶Mn投料比的CsPbCl3∶Mn2+ NCs的TEM图像(标尺为100 nm)和HRTEM图像(a)、XRD谱(b)、XPS谱((c)~(d))以及吸收光谱和光致发光光谱(λex=365 nm) (e);(f)Pb∶Mn=1∶1.0时,不同反应时间下所合成的CsPbCl3∶Mn2+ NCs的光致发光光谱Fig.1 TEM images(the scale bar is 100 nm) and HRTEM images(a), XRD pattern(b), XPS patterns((c)-(d)) and photoluminescence spectra(e) of CsPbCl3∶Mn2+ NCs with different Pb∶Mn feeding ratios synthesized by one-step method during PbCl2 quantification. (f)Photoluminescence spectra of CsPbCl3∶Mn2+ NCs synthesized by rough control method at different reaction time when Pb∶Mn=1∶1.0
另外对所得钙钛矿纳米晶的发光性质进行了表征。图1(e)为所合成的CsPbCl3∶Mn2+纳米晶溶液的吸收光谱和在365 nm激发光下的光致发光光谱,所有样品在365 nm处具有相同的吸光度。可以看出,随着Mn2+投料量的增加,Mn2+与CsPbCl3合金化,使得纳米晶尺寸减小,带隙增大,激子吸收峰逐渐蓝移。合成的CsPbCl3∶Mn2+NCs除了CsPbCl3固有的400 nm的激子发光峰外,均出现了610 nm左右的橙光发射,这来源于Mn2+的自旋禁止的4T1g→6A1gd-d跃迁[26-27]。但是,610 nm的发射峰强度明显高于400 nm的发射峰,而且610 nm的发光峰强度随着Mn2+投料量的增加先升高后降低,在Pb∶Mn=1∶1.0时达到最高,之后强度下降主要是由于Mn晶体场中交叉弛豫引起的非辐射跃迁[29]。我们还探究了反应时间对反应过程的影响,图1(f)为Pb∶Mn=1∶1.0时所得样品随时间变化的光致发光图像。反应时间为0 h时所得样品的PL强度已经是最大值,说明该反应过程速率极快,不易控制,在注入油酸铯前驱体一瞬间即可完成反应,增加反应时间反而不利于该反应过程,只能对Mn2+的掺杂量进行大范围调控。
为了对Mn2+的掺杂含量进行更精细的调控,控制反应速率,我们采用两步热注射法在相同Pb∶Mn投料比的情况下合成了CsPbCl3∶Mn2+NCs。图2(a)为所得纳米晶的TEM图像,可以看出随着Mn2+投料量的增加,纳米晶的尺寸减小,说明Mn2+成功掺入;但在相同Pb∶Mn投料比下,两步法所得纳米晶的尺寸略大于一步法所得纳米晶。进一步对两步法所得纳米晶的发光性质进行了研究,其PL光谱如图2(b)所示,与一步法所得纳米晶相同,均含有400 nm的激子发射峰和610 nm的Mn2+特征峰,且随着Mn2+投料量的增加,400 nm的发射峰强度增强,但610 nm处Mn2+的特征峰强度明显较低。对Pb∶Mn=1∶0和Pb∶Mn=1∶0.5的样品分别测试其发光寿命,如图2(c)、(d)所示,使用三指数衰减函数拟合本征激子发光和Mn2+特征发光的寿命(LT)衰减曲线,发现掺杂Mn2+后本征激子发光寿命下降(从33.5 ns下降到22.8 ns)。这也归因于Mn2+掺杂使纳米晶的光载流子复合路径发生改变,部分能量从宿主激子转移到Mn2+的掺杂能级上,从而加速了激子复合过程;另外,由于Mn2+离子的4T1g和6A1g电子态之间的自旋禁止跃迁,掺杂发光的寿命比本征激子发光的寿命长得多(0.637 msvs33.5 ns)。此外,对一步法和两步法所得样品的光致发光量子产率进行比较。图2(e)为纳米晶的PLQY曲线,与一步法相比,两步法所得纳米晶的PLQY略低,但仍能大幅提升CsPbCl3NCs的PLQY。这主要是因为Mn2+的掺杂引入了掺杂能级,激发到导带的电子会部分转移到掺杂能级,引起掺杂发光,进而减少非辐射复合消耗的电子数的缘故。以上现象与纳米晶中实际Mn2+含量也密切相关,因此我们通过ICP对CsPbCl3∶Mn2+NCs中Mn2+的实际掺杂含量进行测定,如图2(f)所示。在相同Pb∶Mn投料比的情况下,一步法比两步法所得到的样品纳米晶中具有更高的Mn含量,且实际Mn2+含量与Mn2+投料量成正比。这合理解释了与一步法所得纳米晶相比,两步法所得纳米晶尺寸更大,610 nm的发射峰强度更弱;也进一步表明一步法是合金化过程,引入Mn2+较多,而两步法是掺杂过程,只引入微量Mn2+。图2(g)为所得纳米晶溶液在紫外灯下的图像。两步法所得纳米晶溶液能够基本维持CsPbCl3NCs原有的发光颜色;而一步法在开始引入Mn2+时所得纳米晶溶液就已经出现了Mn2+相关的明亮的橙色发光,掩盖了CsPbCl3NCs在400 nm处的本征激子发光。这与两种方法的实际Mn2+含量相一致。以上实验均是在实验体系中PbCl2定量的情况下进行的,通过单独调控MnCl2的投料量即可实现对Mn2+掺杂量的调控。

图2 PbCl2定量时,(a)两步法合成的不同Pb∶Mn投料比的CsPbCl3∶Mn2+ NCs的TEM图像(标尺为100 nm),(b)两步法所合成的不同Pb∶Mn投料比的CsPbCl3∶Mn2+ NCs的光致发光光谱(插图为放大图),(c)两步法合成的Pb∶Mn=1∶0和Pb∶Mn=1∶0.5的CsPbCl3∶Mn2+ NCs的本征激子发光寿命图谱,(d)两步法合成Pb∶Mn=1∶0.5的CsPbCl3∶Mn2+ NCs的Mn2+特征发光寿命图谱;一步法和两步法合成的CsPbCl3∶Mn2+ NCs的PLQY(e)、实际Mn含量与Pb∶Mn投料比的关系曲线(f)、正己烷溶液荧光照片(365 nm激发)(g)及两种不同的形成机制示意图(h)Fig.2 (a)TEM images(the scale bar is 100 nm) of CsPbCl3∶Mn2+ NCs with different Pb∶Mn feeding ratios synthesized by twostep method during PbCl2 quantification. (b)Photoluminescence spectra of CsPbCl3∶Mn2+ NCs with different Pb∶Mn feeding ratios synthesized by two-step method during PbCl2 quantification(inset is amplification of spectra). (c)Intrinsic exciton luminescence lifetime of CsPbCl3∶Mn2+ NCs synthesized by two-step method with Pb∶Mn=1∶0 and Pb∶Mn=1∶0.5. (d)The characteristic luminescence lifetime of Mn2+ of CsPbCl3∶Mn2+ NCs synthesized by two-step method with Pb∶Mn=1∶0.5. PLQY(e), the relationship between the real Mn content and the Pb∶Mn feeding ratio(f), PL image of n-hexane solution NCs under 365 nm irradiation(g), and schematic illustration of mechanisms(h) of CsPbCl3∶Mn2+ NCs synthesized by one-step and two-step methods
以上结果表明,在相同Pb∶Mn投料量下Mn2+的实际掺杂量存在巨大差异,两步法可以更加精确调控Mn2+的掺杂含量。因此,我们认为一步法和两步法在调控CsPbCl3∶Mn2+NCs中Mn2+的实际掺杂含量时存在两种反应过程(如图2(h)所示)。采用一步法进行粗略调控时,PbCl2和MnCl2同时存在于反应溶液中,Pb2+和Mn2+同时形成一定数目的[PbCl6]4-和[MnCl6]4-,在高温下和Cs+直接结合形成CsPbCl3∶Mn2+NCs。采用两步法进行精细调控时,最初反应体系中只存在PbCl2,先合成CsPb-Cl3NCs,之后再单独注入MnCl2的前驱体溶液,此时新加入的Mn2+要取代晶格中的Pb2+,就需要克服Pb与晶格中周围Cs、Cl之间的结合力,将[Pb-Cl6]4-八面体打开[33-34],所以Mn2+的掺杂会更加困难。因此在Pb∶Mn投料比相同的情况下,一步法合成的纳米晶比两步法合成的纳米晶实际Mn2+掺杂量更高,第二种方案更容易达到精细调控CsPbCl3NCs中实际Mn2+掺杂量的目的。
同时,我们采用两步法先合成CsMnCl3NCs,再注入PbCl2前驱体溶液。由图3(a)的光致发光光谱可以看出,刚合成的CsPbCl3∶Mn NCs溶液在Pb含量较少时仅显示400 nm处的激子发射峰,610 nm与Mn相关的发射峰随着Pb含量增加才慢慢显现,但仍然强度较低,最终400 nm的光占主导。这说明此时仍然可以实现对Mn2+的精细调控。两步法中向先合成的CsMnCl3中加入Pb2+,除了要克服Mn2+与周围离子之间的形成能外,Pb2+的半径较大,容易诱发正应力畸变,导致形成能提高,使反应不易进行,从而可以达到精细调控的目的。我们对其晶体结构进行了表征,图3(b)为两步法所得样品的XRD图像,随着Pb-Cl2注射量的增加,纳米晶由CsMnCl3相逐渐转变为纯CsPbCl3相,与一步法明显不同的是两步法合成的纳米晶没有出现CsMnCl3的衍射峰,Mn2+的加入没有改变纳米晶的晶体结构,证明此时合成的纳米晶并不是合金结构,而是Mn2+掺杂的CsPbCl3NCs。

图3 MnCl2定量时,两步法所得纳米晶的光致发光光谱(a)及XRD谱(b)Fig.3 Photoluminescence spectra(a) and XRD pattern(b) of NCs synthesized by two-step method during MnCl2 quantification
3.2 CsPbCl3∶Mn NCs的稳定性
对所得样品进行了稳定性分析,图4(a)为一步法进行粗调时所得纳米晶在室温下存放四周后的TEM图像。纯CsPbCl3相对水氧极不稳定,放置四周后已有部分样品解离;而少量Mn2+合金化后的样品形貌无明显变化,Mn2+过多时会导致钙钛矿加速解离,不利于钙钛矿稳定性的提升。放置四周后样品的XRD谱显示Pb∶Mn投料比为1∶0.5和1∶1.0时钙钛矿晶相相对稳定(如图4(b)所示)。图4(c)为相对应的PL光谱与刚合成样品的PL光谱对比,与刚合成的样品相比,四周后样品的发射峰强度均降低。Pb∶Mn=1∶1.0的样品610 nm处的发射峰强度明显降低,与Pb∶Mn=1∶0.5的样品610 nm处的发光强度一致,而Pb∶Mn=1∶2.0的样品放置四周后不再发光;同时,四周后样品的发射峰位置发生一定程度的蓝移,可能是四周后Mn2+析出,Mn2+-Mn2+对数目减少所致[35]。另外,对两步法合成的掺杂结构进行稳定性分析,以Pb∶Mn=1∶1.0时的样品为例,图4(d)为所得纳米晶在室温下放置两周后的TEM图像。两周后该样品纳米晶尺寸明显增大,且已有CsMnCl3相析出;同时,400 nm的激子发射峰和600 nm的与Mn相关的发射峰强度均明显降低,Pb∶Mn=1∶0.5与Pb∶Mn=1∶1.5时的样品放置两周后Mn2+的特征峰消失,Pb∶Mn=1∶2.0时的样品两周后不再发光。无论是合金结构的纳米晶还是掺杂结构的纳米晶,放置一段时间后双峰强度均降低,但是合金结构的纳米晶本征激子发射峰强度比掺杂结构降低较少。以上结果表明,进入钙钛矿晶格中的Mn2+可以有效地提高钙钛矿的稳定性。这主要是由于适量的Mn2+引入后会导致晶格收缩,使纳米晶形成能提高;而且Mn2+的来源是MnCl2,会额外引入Cl-,减少卤素空位,钝化纳米晶原有的表面缺陷,从而抑制非辐射复合的发生。但是,过量的Mn2+容易破坏钙钛矿晶格,加速钙钛矿解离。此外,一步法形成的合金结构对于钙钛矿稳定性的提升比两步法合成的掺杂结构更加有利,这可能是由于两步法合成时Mn2+进入钙钛矿晶格需要打开[PbCl6]4-八面体对钙钛矿晶格本身产生了轻微破坏所导致的。
3.3 CsPb(ClxBr3-x)∶Mn2+ NCs的合成及发光性能
根据以上研究结果可知,一步法热注射法可以对Mn2+的实际掺杂含量进行大范围调控,合成的纳米晶中Mn2+含量更高,且发光性能更好,在此基础上研究了卤素离子对发光性能的影响。图5(a)为CsPb(ClxBr3-x)∶Mn2+NCs的XRD谱,随着Br-的增加,纳米晶由CsPbCl3相逐渐向CsPbBr3相转变。图5(b)为CsPb(ClxBr3-x)∶Mn2+NCs光致发光光谱以及在紫外灯照射下的溶液图像。通过观察我们可以看到,随着Br-含量由0%逐渐增加到100%,Br-和Cl-交换导致CsPbX3的激子发射峰逐渐红移。然而,当Br-含量在0~50%时,存在Mn2+的特征峰;当Br-含量达到50%后,Mn2+的特征峰消失。这主要是由于CsPbBr3的能带结构与Mn2+的4T1-6A1能级不匹配,因此在Br-含量较高的情况下不会出现Mn2+的特征峰。当Br-含量较低时,体系中以CsPbCl3为主要组成,大半径的Br-的少量加入可以使铅卤八面体打开一个较大的空位,为Mn2+的掺杂提供了条件,从而使Mn的特征峰得以显现。
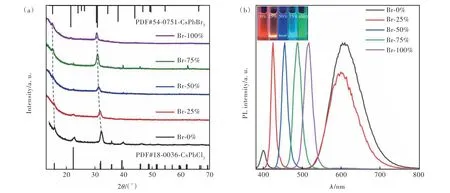
图5 Pb∶Mn=1∶1.0时,采用一步法热注射法合成的不同Cl∶Br投料比的CsPb(ClxBr3-x)∶Mn2+ NCs的XRD图像(a)和光致发光光谱(b),插图为样品的正己烷溶液在365 nm紫外灯照射下的图片Fig.5 XRD pattern(a) and photoluminescence spectra(b) of CsPb(ClxBr3-x)∶Mn NCs with different Cl∶Br feeding ratios synthesized by one-step hot injection method during Pb∶Mn = 1∶1.0. Inset: colloidal solution in n-hexane under UV light(λ = 365 nm)
4 结论
本文揭示了CsPbCl3钙钛矿纳米晶中Mn2+的可控掺杂途径,通过控制反应体系中的Pb∶Mn投料比,采用一步法和两步法对Mn2+的掺杂量进行了粗略和精细调控,有效控制Mn2+的掺杂速率,形成了合金和掺杂结构,分别将CsPbCl3钙钛矿纳米晶的光致发光量子产率由3%提升到77%和66%,同时显著提升了钙钛矿纳米晶结构和发光的稳定性。进一步阐述了一步法和两步法中CsPbCl3∶Mn2+NCs的形成过程,为Mn2+的可控掺杂提供了理论基础,解决了Mn2+掺杂速率过快的问题。最后,通过对卤素离子的调控,将CsPb-(ClxBr3-x)∶Mn2+NCs的发光颜色从蓝光调至橙光,实现了发射光谱的可调性,为CsPbX3钙钛矿纳米晶的显示应用奠定了理论基础。
本文专家审稿意见及作者回复内容的下载地址:http://cjl.lightpublishing.cn/thesisDetails#10.37188/CJL.20230278.
猜你喜欢
当代化工研究(2023年16期)2023-09-11
中国烟草学报(2021年4期)2021-09-26
水泵技术(2021年4期)2021-01-22
纺织科学研究(2020年4期)2020-04-30
物理学报(2019年10期)2019-06-04
山东煤炭科技(2018年1期)2018-12-05
科学之谜(2018年3期)2018-04-09
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10
原子与分子物理学报(2015年1期)2015-11-24
