
吡唑并[1,5-a]嘧啶衍生物的合成研究
2024-03-20 07:33包安丽
山西化工 2024年2期
刘 杨,包安丽,李 娟
(1.四川化工职业技术学院,四川 泸州 646300;2.成都新恒创药业有限公司,四川 成都 611137;3.四川佳乐酒业浓香酿酒有限公司,四川 泸州 646300)
随着药物合成技术的不断发展,在人类多种疾病的治疗中,都能有含氮杂环类药物的存在。吡唑并[1,5-a] 嘧啶类化合物就是含氮杂环类化合物中典型的一类,因其分子结构中同时包含了吡唑与嘧啶两个重要的含氮活性单元,其衍生出的系列取代化合物大多表现出良好的生物活性。在中枢神经系统疾病治疗、抗病毒、抗肿瘤及心血管疾病防治等诸多领域表现突出。这类吡唑并[1,5-a]嘧啶衍生化合物在新型药物中间体及药物分子开发中,表现出极大的潜力,甚至在农药、食品添加剂等领域也有应用前景。
以吡唑并[1,5-a]嘧啶环为母核的衍生化合物,对人体内的许多重要生物靶点都能起作用。如常见的环氧化酶(COX-2)、钾离子通道、外周型苯二氮卓受体及γ-氨基丁酸受体等。针对这些受体已开发并应用的药物很多,如针对入睡困难的镇静催眠药物扎来普隆;新型2-型糖尿病的二肽基肽酶(DPP-4)抑制剂阿拉格列汀等。
此外,吡唑并[1,5-a]嘧啶类衍生化合物在更多其他治疗上的应用也不断被发现。如作为GPR4 受体拮抗剂,用于自身免疫系统相关疾病、炎症性疼痛以及血管生成有关疾病的治疗;作为TRPC6 激活剂或阻断剂,可用于预防和治疗TRPC6 表达异常而引起的心急肥大症及肾小球疾病;作为Cdk4/6 抑制剂,用于抑制乳腺癌细胞和肝癌细胞生长。许多衍生化合物还表现出抗氧化活性、抑菌活性等。
1 合成方法及进展
到目前为止,针对吡唑并[1,5-a]嘧啶类衍生物的合成主要为以下几类方法:
用1,3-二羰基类化合物或α,β-不饱和化合物与氨基吡唑直接缩合;由1H-5-氨基吡唑与不饱和羰基化合物反应合成;以丙二腈、醋酸酐、原甲酸三乙酯为原料,与三氯氧磷反应合成;联烯酮与3-氨基吡唑缩合;常规加热结合微波辐射技术合成7-芳胺基-2-苯基吡唑并[1,5-a]嘧啶;常温催化反应合成,主要以四氢呋喃作溶剂,加入反应物和氢氧化锂水溶液,室温下催化反应。
吡唑并[1,5-a]嘧啶类化合物传统的合成方法较多,但也存在诸多限制。本文实验设计在结合传统合成方法优势的前提下,探索吡唑并[1,5-a]嘧啶类化合物新的合成途径和方法,并合成了多个以吡唑并[1,5-a]嘧啶为母核衍生出的新化合物。
2 实验部分
目标化合物合成路线如图1,以苯甲酰乙腈为起始原料,关环合成3-苯基-5-氨基吡唑,再与4-氯乙酰乙酸乙酯反应合成中间体Ⅰ,加入三氯氧磷取代生成中间体Ⅱ,之后以中间体Ⅱ为母核在5 位氯和7位氯上用不同基团取代合成不同的吡唑并[1,5-a]嘧啶衍生物。此合成路线路径较长,但可以合成关键结构中间体Ⅱ,之后以其为母核容易在5 位和7 位接入不同基团,得到多个衍生化合物。

图1 目标化合物合成路线
2.1 合成原料3-苯基-5-氨基吡唑
以75~100 mL 乙醇作为溶剂,加入0.1 mol 苯甲酰乙腈至烧瓶中,搅拌溶解。再加入0.2 mol 质量分数80%的水合肼,继续搅拌片刻,油浴加热回流反应,温度95 ℃,反应时间6~8 h。反应液先旋干其乙醇溶剂,旋干产物加水搅拌,抽滤,石油醚多次洗涤(或重结晶)。抽干后取固体,烘干即得产物,为灰白色粉末状固体。本步骤收率75%~80%。反应监测时,TLC 展开剂为m(乙酸乙酯)∶m(石油醚)=1∶1。
2.2 合成2-苯基-5-氯甲基-7-羟基吡唑并[1,5-a]嘧啶(中间体Ⅰ)
烧瓶中加入15~20 mL 冰乙酸作为溶剂。搅拌下加入13.5 mmol 3-苯基-5-氨基吡唑,溶解后再加入15 mmol 4-氯乙酰乙酸乙酯。溶解后移至油浴锅中加热回流反应,反应温度110 ℃。4 h 后反应液中有粉红色固体粉末析出,该固体即为中间体Ⅰ产物。中间体Ⅰ不溶于水,加入适量水至烧瓶中,搅拌使反应液中固体分散,抽滤,固体用水洗涤3~5 次,抽干后烘干即得目标产物2-苯基-5-氯甲基-7-羟基吡唑并[1,5-a]嘧啶(中间体Ⅰ),为粉红色晶状粉末。本步骤收率80%~85%。反应监测时,TLC 展开剂为m(甲醇)∶m(二氯甲烷)=1∶10。
2.3 合成2-苯基-5-氯甲基-7-氯吡唑并[1,5-a]嘧啶(中间体Ⅱ)
烧瓶中加入5~10 mL 乙腈作为溶剂。搅拌下加入5 mmol 中间体Ⅰ,溶解后先加入1 mL 吡啶,再缓慢滴加15 mmol 三氯氧磷,升温回流反应,温度110 ℃。反应1 h。将反应液缓慢倒入宽敞烧杯中,倾倒同时加入冰水,反应液放热产生白雾。待反应液全部倒入烧杯后,再加入适量冰水搅拌,用50%氢氧化钠调节pH至中性。分液,加入适量乙酸乙酯萃取,浓缩干燥萃取萃取的乙酸乙酯溶液即得目标产物2-苯基-5-氯甲基-7-氯吡唑并[1,5-a]嘧啶(中间体Ⅱ),本步骤收率56%~58%。反应监测TLC 展开剂为m(乙酸乙酯)∶m(石油醚)=1∶4。
2.4 合成2-苯基-5-氯甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(中间体Ⅲ)
烧瓶中加入10 mL 异丙醇作为溶剂。开启搅拌器边搅拌边加入3 mmol 对甲苯胺使其溶解,溶解完全后加入3 mmol 中间体Ⅱ,移至油浴锅升温反应,温度50 ℃,反应3.5 h。反应液中会有固体粉末析出。反应液抽滤取固体,乙醇加热溶解后用石油醚重结晶。重结晶完全后抽滤,即得目标产物2-苯基-5-氯甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(中间体Ⅲ),本步骤收率73%~78%。反应监测时,先取反应液加二氯甲烷溶解稀释,TLC 展开剂为m(乙酸乙酯)∶m(石油醚)=1∶8~1∶4。中间体Ⅲ用氘代二甲基亚砜溶解,一维氢谱数据如图2:1H NMR(600 MHz,DMSO)δ 11.04(s,1H),8.19(d,J=7.4 Hz,2H),7.54(t,J=7.5 Hz,2H),7.49(t,J=7.3 Hz,1H),7.41(d,J=8.1 Hz,2H),7.37(d,J=8.1 Hz,2H),7.12(s,1H),6.45(s,1H),4.84(s,2H),2.39(s,3H)。
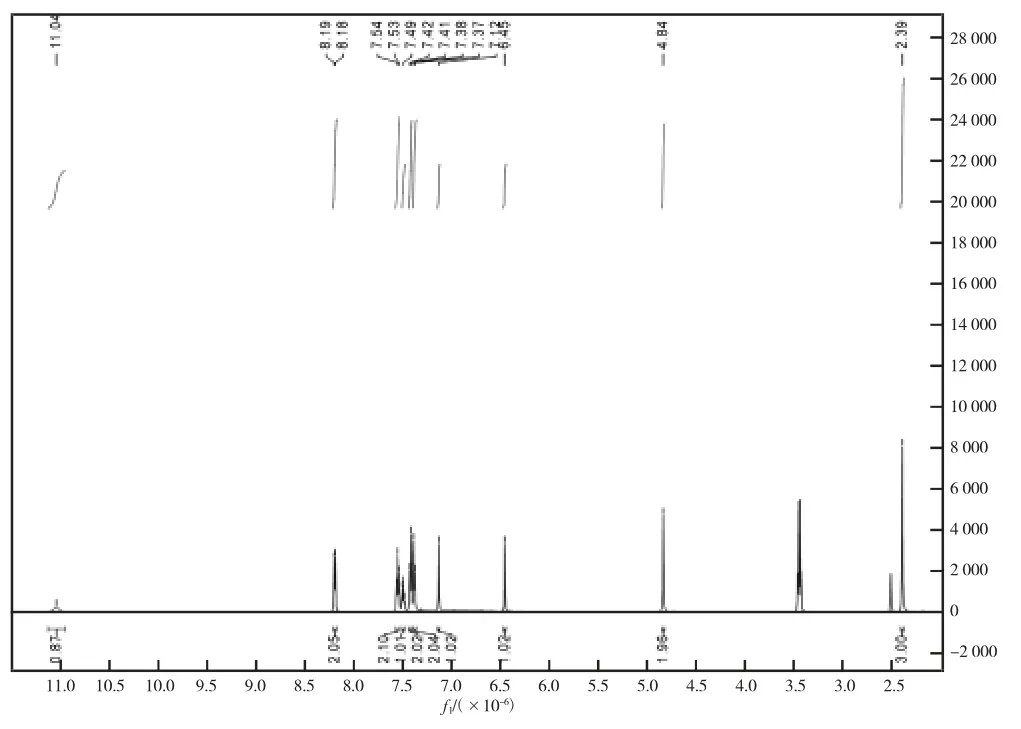
图2 中间体Ⅲ1H NMR 图

图3 化合物011H NMR 图
2.5 合成2-苯基-5-(2-甲基咪唑)甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(化合物01)
烧瓶中加入10 mL 四氢呋喃作为溶剂。先加入6 mmol 叔丁醇钾搅拌使其尽量溶解,之后缓慢加入3 mmol 2-甲基咪唑,室温搅拌20 min 后再加入中间体Ⅲ。油浴加热升温至75 ℃,反应1 h。取反应液减压旋干除去溶剂,用5%HCl 调pH 后加水和乙酸乙酯萃取,分液,取有机相旋干粗硅胶拌样后硅胶柱层析[m(洗脱液甲醇)∶m(二氯甲烷)=1∶40]即得目标产物2-苯基-5-(2-甲基咪唑)甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(化合物01),本步骤收率35%~40%。反应监测时,取反应液先调pH 加乙酸乙酯萃取,TLC 展开剂m(甲醇)∶m(二氯甲烷)=1∶20。
化合物01 用氘代氯仿溶解,一维氢谱数据:
1H NMR(600 MHz,CDCl3)δ 8.22(s,1H),8.02(d,J=7.3 Hz,2H),7.50(t,J=7.6 Hz,2H),7.43(t,J=7.3 Hz,1H),7.25(d,J=8.1 Hz,2H),7.15(d,J=8.2 Hz,2H),6.98(s,1H),6.92(s,1H),6.83(s,1H),5.83(s,1H),5.07(s,2H),2.40(s,3H),2.38(s,3H)。
2.6 合成2-苯基-5-三氮唑甲基-7-三氮唑基吡唑并[1,5-a]嘧啶(化合物02)
烧瓶中加入10 mL 二甲亚砜作为溶剂。边搅拌边加入10 mmol 1,2,4-三氮唑,接着加入2.5 mmol 碳酸钾,搅拌20 min 后加入中间体Ⅱ,移至油浴加热回流反应,温度120 ℃,反应3.5 h。产物处理:反应液加水搅拌,在加入乙酸乙酯萃取。取乙酸乙酯层真空旋干加粗硅胶拌样。因溶剂为二甲基亚砜,萃取液中含少量二甲基亚砜无法旋干,硅胶柱层析[洗脱液m(甲醇)∶m(DCM)=1∶10]分离纯化。过柱后产物加水析出淡黄色固体。抽滤并用石油醚洗涤,减压旋干即得目标产物2-苯基-5-三氮唑甲基-7-三氮唑基吡唑并[1,5-a]嘧啶(化合物02),本步骤收率18%~24%。反应监测时,取反应液于离心管中先加水,再用乙酸乙酯萃取,TLC 展开剂m(甲醇)∶m(二氯甲烷)=1∶10。
化合物02 用氘代二甲基亚砜溶解,一维氢谱图谱如图4,数据如下:1H NMR(600 MHz,DMSO)δ 10.35(s,1H),9.06(s,1H),8.71(s,1H),8.61(s,1H),8.26(s,1H),8.20(s,1H),8.17(d,J=7.9 Hz,3H),8.03(s,1H),7.91(d,J=5.3 Hz,4H),7.53 7.49(m,5H),7.31(s,1H),6.80(s,1H),5.69(s,2H)。

图4 化合物021H NMR 图
2.7 合成2-苯基-5-对甲苯胺甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(化合物03)
烧瓶中加入10 mL 异丙醇作为溶剂。开启搅拌器边搅拌边加入2.6mmol 对甲苯胺,搅拌溶解完全后再加入2mmol 中间体Ⅱ和适量碳酸钾,升温回流反应,温度90 ℃。反应2 h。反应液减压旋干溶剂异丙醇,之后加水除去反应液中的碳酸钾,过滤取固体,加乙酸乙酯溶解后用石油醚重结晶,硅胶柱层析[洗脱液m(乙酸乙酯)∶m(石油醚)=1∶10~1∶8]得目标产物2-苯基-5-对甲苯胺甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(化合物03),本步收率38%~43%。反应监测时,取反应液于离心管中加乙酸乙酯稀释,TLC监测展开剂m(乙酸乙酯)∶m(石油醚)=1∶4。
化合物03 用氘代氯仿溶解,一维氢谱图谱如图5,数据如下:1H NMR(600 MHz,CDCl3)δ 8.11(s,1H),8.01(d,J=7.0 Hz,2H),7.47(t,J=7.5 Hz,2H),7.40(t,J=7.4 Hz,1H),7.21(d,J=8.1 Hz,2H),7.17(d,J=8.4 Hz,2H),6.98(d,J=8.5 Hz,2H),6.80(s,1H),6.58(d,J=8.5 Hz,2H),6.34(s,1H),4.32(s,2H),2.38(s,3H),2.24(s,3H)。

图5 化合物031H NMR 图
3 结论
经过多条路线设计探索和大量的实验研究,最终确定以苯甲酰乙腈为起始原料,先关环合成3-苯基-5-氨基吡唑,再与4-氯乙酰乙酸乙酯合成7-羟基-5-氯甲基-2-苯基吡唑并[1,5-a]嘧啶(中间体Ⅰ),然后加入三氯氧磷取代生成7-氯-5-氯甲基-2-苯基吡唑并[1,5-a]嘧啶(中间体Ⅱ),之后以7-氯-5-氯甲基-2-苯基吡唑并[1,5-a]嘧啶为母核在5 位氯和7 位氯上可以比较容易地用不同潜在活性基团取代,合成一系列吡唑并[1,5-a]嘧啶衍生物。
本文主要合成了以下3 个新的化合物:2-苯基-5-(2-甲基咪唑)甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(化合物01)、2-苯基-5-三氮唑甲基-7-三氮唑基吡唑并[1,5-a]嘧啶(化合物02)以及2-苯基-5-对甲苯胺甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(化合物03)。此外探索了2-苯基-5-氯甲基-7-对甲苯胺基吡唑并[1,5-a]嘧啶(中间体Ⅲ)新的合成路线。
猜你喜欢
化工设计通讯(2024年1期)2024-04-08
世界农药(2023年8期)2023-09-04
中国药学药品知识仓库(2022年10期)2022-05-29
今日农业(2021年2期)2021-11-27
今日农业(2020年23期)2020-12-31
汕头大学学报(自然科学版)(2020年4期)2020-12-14
合成化学(2015年1期)2016-01-17
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
化工进展(2015年3期)2015-11-11
无机化学学报(2014年12期)2014-02-28
