江汉油田中高温高矿化度油藏采油功能菌群落结构分析
2024-03-20 08:38曲瑞雪牟莎莎邓舒元孙珊珊佘跃惠王正良
化学与生物工程 2024年3期
徐 峰,姚 快,曲瑞雪,牟莎莎,邓舒元,孙珊珊*,佘跃惠,王正良
(1.江汉油田石油工程技术研究院,湖北 武汉 430000;2.非常规油气省部共建协同创新中心,湖北 武汉 430100;3.油气钻采工程湖北省重点实验室,湖北 武汉 430100;4.长江大学石油工程学院,湖北 武汉 430100;5.中国地质大学(北京)能源学院,北京 100083;6.长江大学化学与环境工程学院,湖北 荆州 434000)
稠油是世界原油资源的重要组成部分,与轻质原油相比,其黏度高、流动性差、开采难度较大[1]。江汉油田普通稠油油藏大多属难动用储量,难以实现经济有效的开发。微生物提高原油采收率(microbial enhanced oil recovery,MEOR)技术是一种环境友好、成本较低、高效、可持续作用的方法[2]。油藏是一个寡营养的极端环境,通过向油藏中注入碳源、氮源及磷源等有机或无机营养物质来激活某些内源采油功能菌[3-4],利用内源微生物的繁殖产生代谢物(如小分子有机酸、气体、生物表面活性剂、生物聚合物等),达到乳化原油、降解原油组分、改变储层物性等目的,进而提高采收率[5-6]。由于油藏微生物的种类繁多,针对性激活内源采油功能菌、维持微生物高效可持续作用原油是MEOR技术的关键。目前,针对稠油油藏的微生物生态学研究是解决该关键问题的基础。鉴于此,作者通过二代测序技术对江汉油田稠油油藏4个中高温高矿化度区块采出液中的细菌和古菌群落多样性进行研究,为后续激活中高温油藏内源微生物提高原油采收率技术的发展和应用提供理论支撑。
1 实验
1.1 材料
采出液取自江汉油田的4个中高温区块,分别标记为H2-12、Z-2X、YP-9、W-1X;其中,H2-12和Z-2X取自60~70 ℃油藏,YP-9和W-1X取自50 ℃油藏。将采出液样品放入厌氧瓶中,置于实验室-4 ℃冰箱中冷藏保存,防止与空气接触。
1.2 方法
1.2.1 采出液的离子组成分析
1.2.2 DNA提取与检测
用无菌磷酸缓冲液冲洗样品(去除采出液中的原油组分),抽滤,收集样品中的菌体,提取 DNA,采用琼脂糖凝胶电泳对其中细菌和古菌的群落组成进行检测。检测后样品置于-20 ℃下保存[8],进行后续分析。
1.2.3 PCR 扩增[9]
采用引物对 338F、806R对细菌16S rRNA基因V3-V4区进行PCR扩增;采用引物对519F(5′-CAGCCGCCGCGGTAA-3′)、915R(5′-GTGCTCCCCCGCCAATTCCT-3′)对古菌16S rRNA基因V4-V5区进行PCR扩增。PCR体系为:Q5 high-fidelity DNA polymerase 0.25 μL,5×Reaction Buffer 5 μL,dNTP(10 mmol·L-1)2 μL,模板 DNA 2 μL,正向引物(10 μmol·L-1)1 μL,反向引物(10 μmol·L-1)1 μL,水 8.75 μL。扩增程序为:94 ℃预变性5 min;94 ℃变性1 min,52 ℃退火1 min,72 ℃延伸10 min,保持4 ℃,30个循环。前引物中的barcode是7个碱基的寡核苷酸序列,用来区分同一文库中的不同样品。PCR 采用 NEB Q5 DNA 高保真聚合酶,用 2%琼脂糖凝胶对扩增产物进行电泳;切取目的片段,并用 Axygen 凝胶回收试剂盒回收。
在Microplate Reader(BioTek,FLx800)上利用Quant-iT PicoGreen dsDNA Assay Kit对PCR产物进行定量,然后将每个样品按照其所需的数量进行混合。
1.2.4 文库构建
利用Illumina公司的TruSeq Nano DNA LT Library Prep Kit构建文库。首先利用试剂盒中的EndRepair Mix2将DNA 5′端突出的碱基切除,然后将3′端缺失的碱基补齐,同时在5′端加上1个磷酸基团,进行末端修复。
2 结果与讨论
2.1 采出液的离子组成
4种样品的离子组成如表1所示。
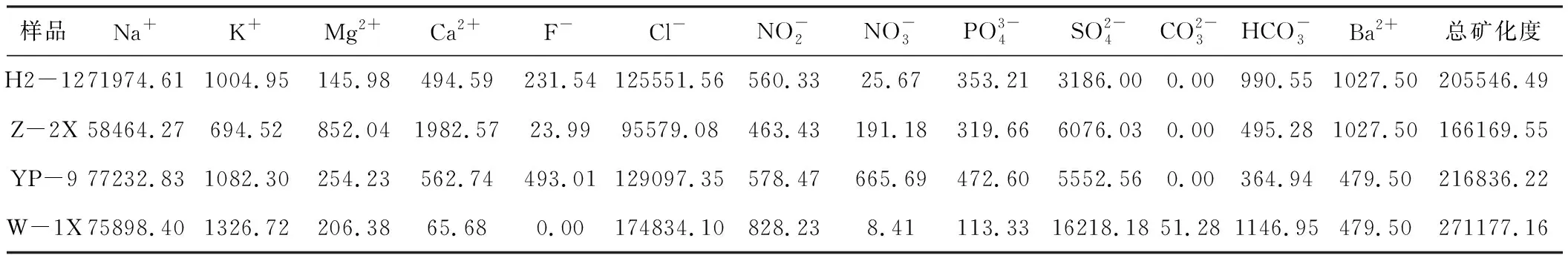
表1 4种样品的离子组成/(mg·L-1)
2.2 细菌群落组成分析
2.2.1 物种分类学注释
采用QIIME2的classify-sklearn算法[10](https://github.com/QIIME2/q2-feature-classifier),具体步骤如下:对于每个ASV(扩散序列变异)的特征序列或每个OTU(操作分类单元)的代表序列,在QIIME2软件中使用默认参数,使用预先训练好的Naive Bayes分类器进行物种注释,即与参考序列数据库进行比对,根据比对结果进行打分判定。通过16S rDNA测序,对4种样品中的细菌进行分类学统计及多样性揭示,物种分类学注释结果如图1所示。
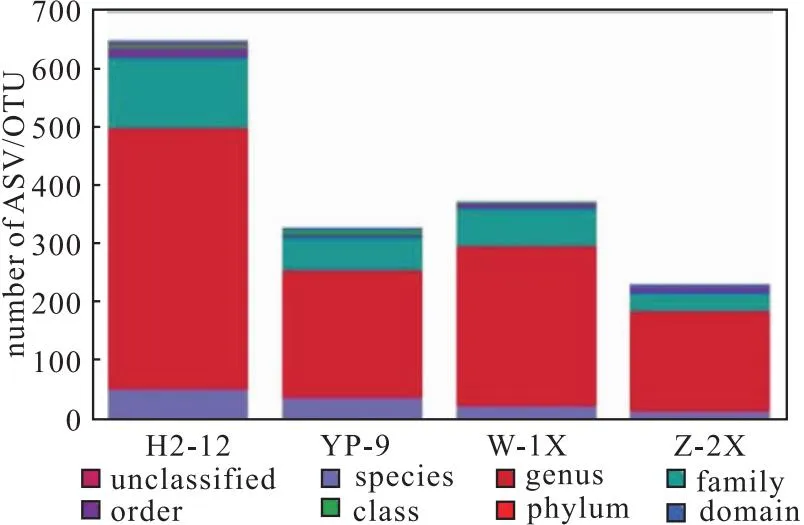
图1 4种样品中细菌的分类学注释Fig.1 Taxonomic notes on bacteria in four samples
从图1可知,H2-12样品中细菌种类最多,其次是W-1X样品,Z-2X样品中细菌种类最少,不足H2-12样品中的一半。
2.2.2 Alpha多样性分析
Alpha多样性指在一个生态系统内或者特定区域内反映物种丰度和均匀度的综合指标[11]。4种样品中细菌的稀疏曲线如图 2 所示。
从图2可知,随着测序深度的增加,4种样品中细菌群落的ASV/OTU总数均先增加后逐渐趋于稳定,说明测序深度合适、取样合理。4种样品中细菌群落的Alpha 多样性指数如表2所示。
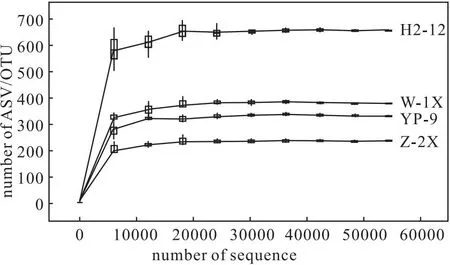
图2 4种样品中细菌的稀疏曲线Fig.2 Sparse curves of bacteria in four samples
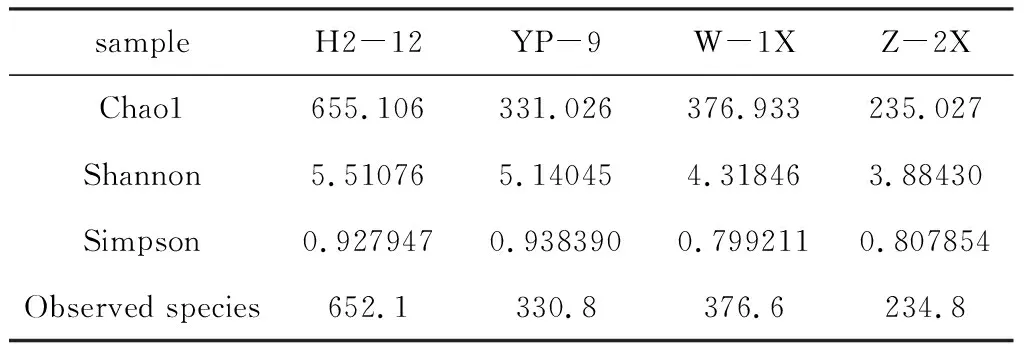
表2 4种样品中细菌群落的Alpha多样性指数
Chao1和Observed species指数表征丰度,Shannon和Simpson指数表征多样性[12]。从表2可知,H2-12样品的Chao1、Shannon和Observed species指数均最高,分别为655.106、5.510 76和652.1,其Simpson指数为 0.927 947,仅次于YP-9样品的Simpson指数(0.938 390),表明H2-12样品的物种丰度和多样性均最高;Z-2X样品的Chao1、Shannon和Observed species指数均最低,表明Z-2X样品的物种丰度和多样性均最低,这与物种分类学注释结果一致。
2.2.3 Beta多样性分析
Beta多样性聚类分析多采用层次聚类(hierarchical clustering)法,以等级树的形式展示样本间的相似度,通过聚类树的分枝长度衡量聚类效果。聚类分析可以采用任何距离评价样本之间的相似度。 4种样品中细菌群落在属水平上的层次聚类分析结果如图 3所示。
从图3可知,YP-9和H2-12样品的距离最近,且同为一支,说明YP-9和H2-12样品中细菌的微生物群落最为相似;W-1X 样品属于单独一支,离其它样品较远,说明W-1X样品与其它样品中细菌的微生物群落差距较大。
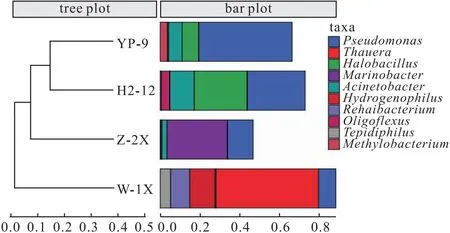
图 3 4种样品中细菌群落在属水平上的层次聚类分析Fig.3 Hierarchical clustering analysis of bacterial communities in four samples at genus level
2.2.4 物种组成分析
高通量测试结果通过BLAST比对后,将4种样品的细菌群落组成在门水平和属水平上进行分类统计,结果如图4所示。

图 4 4种样品在门水平(a)和属水平(b)的细菌群落组成Fig.4 Bacterial community composition of four samples at phylum level(a) and genus level(b)
从图4a可知,在门水平上,Proteobacteria在4种样品中占有绝对优势,占比在53%~96%之间。研究[13-14]发现,Proteobacteria在自然水体、人工水体中均占比较大,在污水处理及氮磷形态转换方面具有十分重要的作用 。H2-12 和 Z-2X 样品中还有较多的Firmicutes(33%、42%)。
从图4b可知,在属水平上,4种样品的细菌群落主要由Pseudomonas(9%~47%)、Thauera(0%~52%)、Halobacillus(0%~27%)、Marinobacter(0%~30%)、Acinetobacter(1%~12%)、Hydrogenophilus(0%~13%)、Rehaibacterium(0%~10%)、Oligoflexus(0%~4%)、Tepidiphilus(0%~5%)和Methylobacterium(0%~4%)组成。
其中,H2-12和YP-9样品中细菌的优势菌群为Pseudomonas(29%、47%)、Halobacillus(27%、8%)和Acinetobacter(12%、7%)。Pseudomonas是革兰氏阴性好氧棒状细菌,能从大多数环境中分离出来,可产鼠李糖脂型表面活性剂,具有降解石油烃、驱油、修复烃类污染、降黏等作用。马鑫等[15]研究发现,Pseudomonasaeruginosa发酵液在模拟实验中的驱油率较水驱提高了33.5%。Halobacillus属于芽孢杆菌科(Bacillaceae),分离自盐湖、盐土、海洋太阳能盐场等高盐环境中,属嗜盐菌。Halobacillusspp.在石油烃类化合物的生物降解中具有重要作用[16]。Acinetobacter具有代谢多样性,能降解己内酰胺、除草剂、有机磷农药和多种石油烃组分等,可产鼠李糖脂型表面活性剂,也可进行反硝化和氨氧化作用。
W-1X样品中细菌的优势菌群为Thauera(52%)、Hydrogenophilus(13%)和Rehaibacterium(10%)。Thauera属于脱氮菌,在反硝化系统运行 51 d以上时,Thauera的相对丰度在 60%以上[17-18]。
Z-2X样品中细菌的优势菌群为Pseudomonas(13%)和Marinobacter(30%)。Marinobacter是目前国外报道较多的一类能够耐高盐环境的反硝化微生物,具有潜在的工业应用价值[19-21]。
2.2.5 代谢通路分析
微生物生态学研究也需要关注菌群所具备的功能潜能,了解检测样本中菌群功能潜能的概况,能最大化扩增子测序性价比高的优势,此外,分析结果或许还能帮助指导后续实验的设计(如宏基因组测序)。获得的功能单元可以依据代谢通路数据库和一定的计算方法获得代谢通路的丰度。4种样品中的细菌代谢通路主要分为生物合成、降解/利用/同化、脱毒、前体代谢物和能量的产生、聚糖途径、高分子修饰和代谢簇。通过对4种样品中细菌群落的代谢通路分析可知,生物合成中丰度最高的是辅因子、辅基、电子载体、维生素生物合成和氨基酸生物合成;降解/利用/同化中丰度最高的是芳香族化合物降解、碳水化合物降解及核苷和核苷酸降解;前体代谢物和能量的产生中丰度最高的是TCA循环和发酵作用。
2.3 古菌群落组成分析
2.3.1 物种分类学注释
通过16S rDNA测序,对4种样品中的古菌进行分类学统计及多样性揭示,物种分类学注释结果如图5所示。
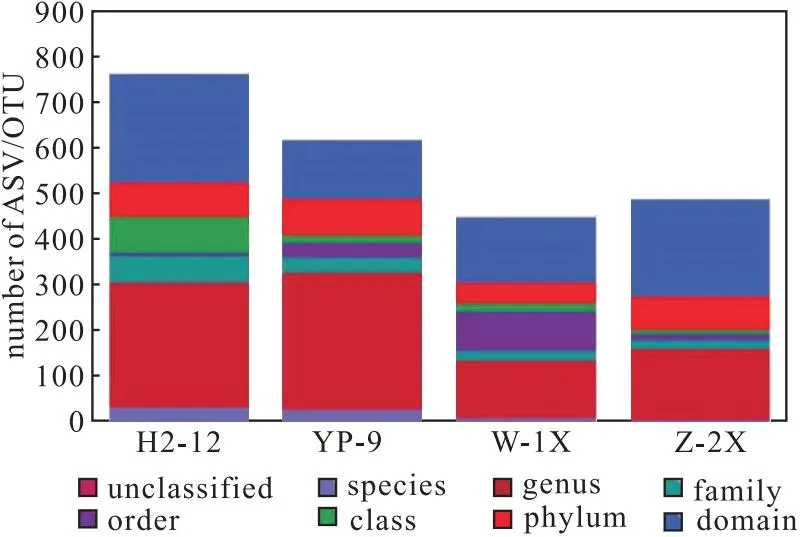
图5 4种样品中古菌的分类学注释Fig.5 Taxonomic notes on archaea in four samples
从图5可知,H2-12样品中古菌种类最多,这与细菌的物种分类学注释结果一致,其次是YP-9样品,W-1X样品中古菌种类最少。4种样品中古菌种类均高于细菌种类,表明样品中古菌种类很丰富。
2.3.2 Alpha多样性分析
4种样品中古菌的稀疏曲线如图6所示。
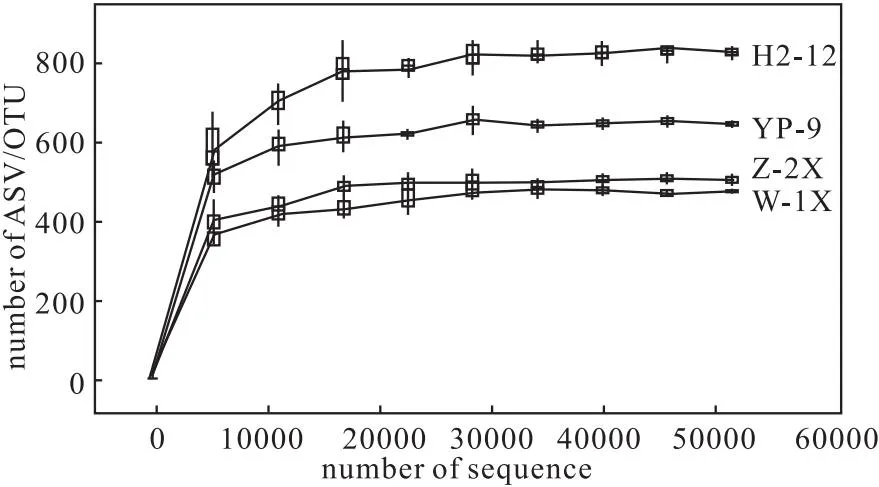
图6 4种样品中古菌的稀疏曲线Fig.6 Sparse curve of archaea in four samples
从图6可知,随着测序深度的增加,4种样品中古菌群落的ASV/OTU总数均先增加后逐渐趋于稳定,说明测序深度合适,取样数量合理,取样数量更多只会产生少量新的OTU。
4种样品中古菌群落的Alpha多样性指数如表3所示。
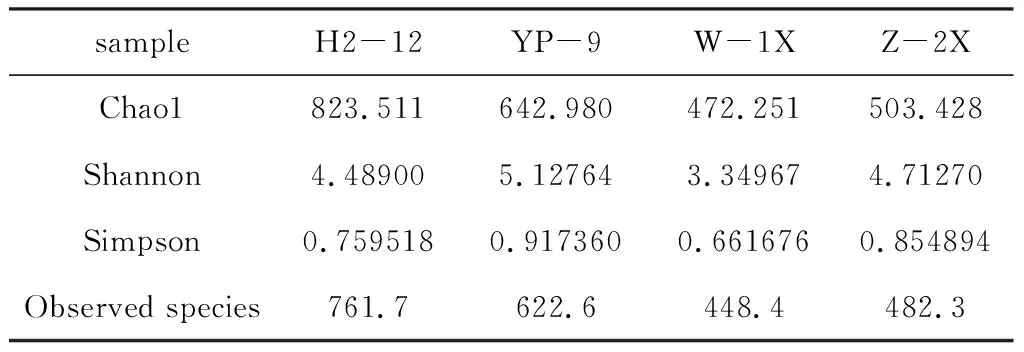
表3 4种样品中古菌群落的Alpha 多样性指数
从表3可知,H2-12样品的物种丰度最高,Chao1和Observed species指数分别为823.511和761.7,这与物种分类学注释结果一致;YP-9样品的物种多样性最高,Shannon和Simpson指数分别为5.127 64和0.917 360;W-1X样品的物种丰度和多样性均最低。根据Chao1和Observed species指数可知,4种样品中古菌群落的丰度均高于细菌群落。
2.3.3 Beta多样性分析
4种样品中古菌群落在属水平上的层次聚类分析结果如图7所示。
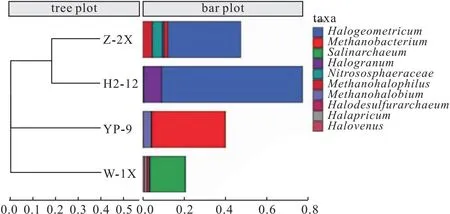
图7 4种样品中古菌群落在属水平上的层次聚类分析Fig.7 Hierarchical clustering analysis of archaea communities in four samples at genus level
从图7可知,Z-2X和H2-12样品的距离最近,且同为一支,说明Z-2X和H2-12样品中古菌的微生物群落最为相似;W-1X样品属于单独一支,离其它样品较远,说明W-1X 样品与其它样品中古菌的微生物群落差距较大。
2.3.4 物种组成分析
4种样品在门水平和属水平的古菌群落组成如图8所示。

图8 4种样品在门水平(a)和属水平(b)的古菌群落组成Fig.8 Archaea community composition of four samples at phylum level(a) and genus level(b)
从图8a可知,在门水平上,4种样品的古菌群落主要以Euryarchaeota(29%~96%)为主;此外,H2-12样品中的优势菌门还有Crenarchaeota(11%),Z-2X样品中的优势菌门还有Thaumarchaeota(5%)。
从图8b可知,在属水平上,4种样品的古菌群落主要由Halogeometricum(0%~69%)、Methanobacterium(0%~36%)、Methanothermobacter(0%~22%)、Salinarchaeum(0%~17%)、Halogranum(0%~9%)、Nitrososphaeraceae(0%~5%)、Methanohalophilus(0%~5%)、Methanohalobium(0%~4%)、Halodesulfurarchaeum(0%~1%)和Halapricum(0%~1%)组成。

W-1X样品中古菌的优势菌群主要为Salinarchaeum(17%)、Halodesulfurarchaeum(1%)和Halapricum(1%)。Salinarchaeum和Halapricum均为嗜盐菌属,能将硝酸盐还原为亚硝酸盐,是一种较强的SRB抑制剂。Halapricum由革兰氏染色阴性的球状或卵球形多形性细胞组成,在含2.5~5.1 mol·L-1NaCl的培养基中生长,最适生长温度为37 ℃。Halodesulfurarchaeum隶属于嗜盐菌纲,由专性厌氧、极端嗜盐的广古菌门组成,以单质硫、二甲基亚砜或硫代硫酸盐为电子受体,通过 H2或甲酸氧化生长。
2.3.5 代谢通路分析
通过对4种样品中古菌群落的代谢通路分析可知,生物合成中丰度最高的是核苷和核苷酸生物合成,其次是氨基酸生物合成;降解/利用/同化中丰度最高的是C1化合物利用与同化;前体代谢物和能量的产生中丰度最高的是TCA循环和发酵作用,这与细菌群落中的前体代谢物和能量的产生丰度是一致的。
3 结论
利用二代测序技术对江汉油田普通稠油油藏的4个采出液样品中的细菌和古菌群落组成进行了多样性分析。物种分类学注释结果表明,H2-12样品中的微生物种类最多,4种样品中古菌种类均高于细菌种类,表明样品中古菌种类很丰富。Alpha多样性分析结果表明,4种样品中古菌群落的丰度均高于细菌群落。在门水平上,细菌群落主要以Proteobacteria(53%~96%)为主;古菌群落主要以Euryarchaeota(29%~96%)为主。在属水平上,细菌的优势菌群主要为Pseudomonas(9%~47%)、Thauera(0%~52%)、Halobacillus(0%~27%)和Marinobacter(0%~30%);古菌的优势菌群主要为Halogeometricum、Methanobacterium和Methanothermobacter。细菌群落组成分析可知,4种样品中的内源微生物群落均具有降黏和脱氮的性能,表明江汉油田内源微生物具有降黏和脱氮的巨大潜力,可通过不同培养基富集筛选高效的降黏和脱氮菌株,进而大大提高江汉油田的原油采收率。
猜你喜欢
东方娱乐周刊(2023年8期)2023-10-03
土壤学报(2022年3期)2022-08-26
大自然探索(2022年5期)2022-07-11
知识就是力量(2022年6期)2022-06-16
课堂内外·教师版(2022年4期)2022-05-23
敦煌学辑刊(2017年2期)2017-11-09
中央民族大学学报(自然科学版)(2016年2期)2016-06-27
新疆大学学报(哲学社会科学版)(2015年6期)2015-10-12
中央民族大学学报(自然科学版)(2015年3期)2015-06-11
应用海洋学学报(2014年4期)2014-11-22