
土壤中多环芳烃衍生物污染特征及迁移转化
2024-03-28 08:11程鹏飞王泽铭赵旭强高彦征江苏大学环境健康与生态安全研究院环境与安全工程学院江苏镇江1013南京农业大学土壤有机污染控制与修复研究所江苏南京10095广东省科学院生态环境与土壤研究所广东省农业环境综合治理重点实验室广东广州510650
中国环境科学 2024年3期
程鹏飞 ,王泽铭 ,赵旭强 ,高彦征 *(1.江苏大学,环境健康与生态安全研究院,环境与安全工程学院,江苏 镇江 1013;.南京农业大学,土壤有机污染控制与修复研究所,江苏 南京 10095;3.广东省科学院生态环境与土壤研究所,广东省农业环境综合治理重点实验室,广东 广州 510650)
多环芳烃(PAHs)衍生物是由PAHs 中氢被其他官能团取代所形成,如氯代、硝基、甲基、氨基和含氧PAHs[1].PAHs 衍生物与PAHs 一样往往由人类活动造成并排放到环境中,但具有更强生物毒性、环境持久性、生物累积性,对食品安全和人体健康存在较大风险[2].受检测手段限制,PAHs 衍生物被发现和研究较晚,现阶段尚未有效监管,属于典型新型持久性有机污染物[3].土壤是持久性有机污染物重要储存库,人类活动产生的PAHs 衍生物通过干湿沉降、灌溉等方式进入土壤[4-5].近年来,土壤PAHs 衍生物污染逐渐受到关注.
氯代多环芳烃(ClPA H s)和硝基多环芳烃(NPAHs)是土壤中常见的两类PAHs 衍生物,商业、工业、交通及住宅用地中均有ClPAHs 和NPAHs检出[6-8].农田土壤中也逐渐检测到低浓度的ClPAHs 和NPAHs,Ni 等[9]在深圳地区农田土壤中检测到ClPAHs(0.33µg/kg),Sun 等[10]则发现我国26个省份农田土壤中普遍存在NPAHs(50µg/kg).农田土壤PAHs 衍生物污染会直接影响食品安全,市售蔬菜和稻米中也检测到ClPAHs(14.7~15.8µg/kg)和NPAHs(ND~4.19µg/kg)[11-12].鉴于ClPAHs 和NPAHs 的持久性和高毒性,土壤中长期低水平暴露会对人体健康产生危害[3].土壤中PAHs 衍生物的赋存形态和生物有效性受其迁移转化行为影响,-Cl 和-NO3为憎水基、亲电子基以及助/生色团,使ClPAHs 和NPAHs 物理、化学和生物特性与母体PAHs 不同,迁移转化机制更多样化[13].因此,开展ClPAHs 和NPAHs 污染和环境行为研究对于环境污染风险评估和防治具有重要意义.近年来,ClPAHs 和NPAHs 在土壤中迁移转化规律已得到广泛关注,开展了吸附-解吸、光降解、氧化还原及微生物降解等基础科学方面研究,但缺少系统的归纳总结.本文对新型污染物PAHs 衍生物在土壤中污染特征和迁移转化规律进行总结,提出未来研究方向和内容,为人们全面认识土壤中PAHs 衍生物迁移转化过程提供参考.
1 土壤中ClPAHs 和NPAHs 污染特征
1.1 土壤中ClPAHs 污染特征
土壤中ClPAHs 污染浓度、组成及分布主要受污染源控制.表1列出国内外土壤中ClPAHs和PAHs的浓度和主要成分.ClPAHs 总浓度范围为0.11~2880µg/kg,比母体PAHs(2200~158000µg/kg)低0~4个数量级.从研究区域看,文献报道的土壤ClPAHs污染主要出现在中国、越南、菲律宾、加纳等亚非国家.电子垃圾拆解是土壤中ClPAHs 重要来源,亚非国家等发展中国家是全球电子垃圾主要集中地,大多采用低技术含量方法拆解.从土壤利用类型分析,ClPAHs 主要分布在典型污染场地如电子垃圾、阻燃剂制造厂、交通道路及周边土壤[14-15]. Wang等[4]在石油化工厂(3.12µg/kg)、阻燃剂制造厂(1.48µg/kg)、电子垃圾回收站(0.26µg/kg)土壤中检测到 8~11 种 ClPAHs,周边土壤也检测到较高ClPAHs(0.48~3.23µg/kg).Nishimura 等[16]在电子废弃物露天焚烧土壤中检测到 26 种 ClPAHs(21~2800µg/kg),其中越南河内电缆电线焚烧厂(2800µg/kg)是目前污染浓度最高的土壤.农田土壤中ClPAHs 浓度相对较低,Ma 等[5]在农田土壤中检测到3-氯荧蒽(3-ClFlt, 0.05µg/kg)和6-氯苯并[a]芘(6-ClBaP, 0.10µg/kg),Ni等[9]则在深圳农业用地中检测到 2-氯蒽(2-ClAnt, ND~0.91µg/kg)、9-氯菲(9-ClPhe, ND~0.51µg/kg)和 9,10-二氯蒽(9,10-DClAnt, ND~0.33µg/kg).从土壤 ClPAHs 组成来看,2-氯蒽(2-ClAnt)、9,10-DClAnt、9-ClPhe、1-氯芘(1-ClPyr)、3-ClFlt、7-氯苯并[a]蒽(7-ClBaA)和 6-ClBaP 是 ClPAHs 主要成分.表 1 显示6-ClBaP(ND~84%)在大多数土壤ClPAHs 占有主要地位,Ma 等[5]发现6-ClBaP 在电子回收站和化工用地土壤ClPAHs 占49%和84%.较低含量苯并[a]芘(BaP, 2.3%~3.9%)对应高含量6-ClBaP,高含量菲(Phe)、芘(Pyr)、荧蒽(Flt)对应 ClPAHs 却低于6-ClBaP,(Chr, 7%~15%)对应氯代(ClChr)未检测到.土壤中6-ClBaP 和BaP 具有相关性,其他ClPAHs 与母体无相关性.6-ClBaP 可由BaP 直接氯化形成,土壤中有较高含量6-ClBaP,而其他ClPAHs很难直接氯化,主要来自二次反应[17]. Xu 等[18]发现PAHs 氯化动力学与其电离能(IP)、分子硬度(η)等电化学性质呈正相关.BaP 比其他PAHs 具有较小IP值和较高反应性,这也可解释6-ClBaP 更可能被直接氯化形成.同样解释土壤中高含量低活性(Chr,IP=7.60)、芴(Flu, IP=7.88)、荧蒽(Flt, IP=7.90)对应ClPAHs 含量却不如高活性蒽(Ant, IP=7.44)、Pyr(IP=7.43) 和 BaP(IP=7.10) 对应 ClPAHs 含量高[5,9,19-20].土壤PAHs 含量与ClPAHs 也会存在明显相关,Nishimura 等[19]发现土壤中 Phe 是(18%~53%)PAHs 主要成分,则检测到种类丰富的高含量ClPhe,如 9-ClPhe(11%~23%)、3,9-二氯菲(3,9-DClPhe, 5.3%~25%)、9,10-二氯菲(9,10-DClPhe,3.4%~19%)、1,9-二氯菲(1,9-DClPhe, ND~0.6%)、3,9,10-三氯菲(3,9,10-TClPhe, ND~1.3%).同种PAH有诸多Cl 代同分异构体,土壤中ClPAHs 中Cl 取代位置与C 原子活性相关,如9,10-DClAnt 中C9 和C10,1-ClPyr 中C1,6-ClBaP 中C6[1].目前,ClPAHs 检测方法尚不完善,以往研究中ClPAHs 种类和浓度的精确度和准确度仍然不足,需优化前处理和检测技术并建立完善的土壤ClPAHs 污染数据库.
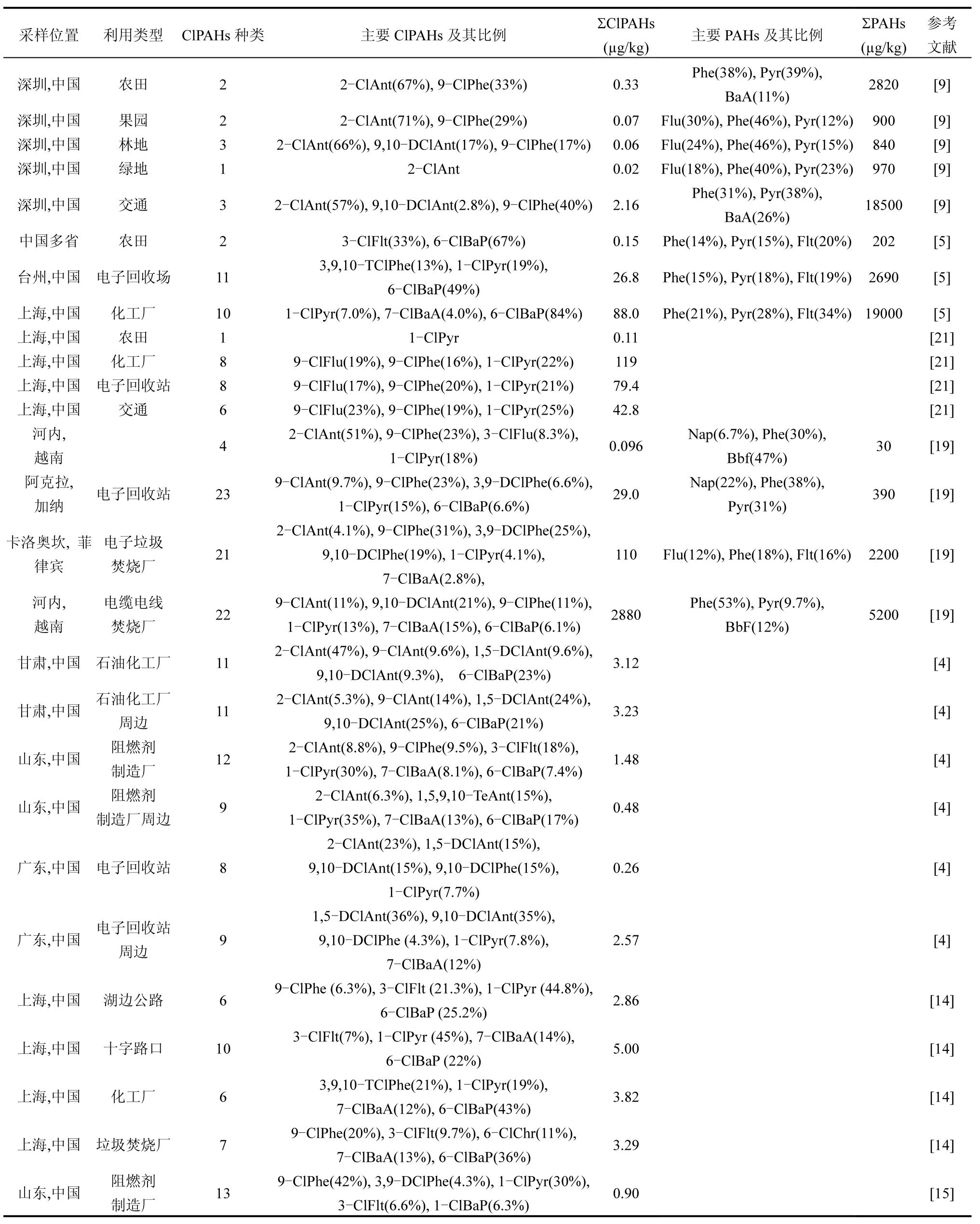
表1 土壤中氯代多环芳烃污染特征Table 1 The contaminant characteristics of ClPAHs in soils
1.2 土壤中NPAHs 污染特征
表2 列出国内外土壤中NPAHs 和PAHs 浓度和主要成分.NPAHs 总浓度为0.184~5280µg/kg,比母体PAHs(81~122876µg/kg)低1~4 个数量级.从研究区域看,NPAHs 分布比ClPAHs 广,中国、澳大利亚、西班牙、瑞士等地都有研究.NPAHs 来源比ClPAHs 更普遍,可从柴油等燃烧源排放,也可由PAHs 与OH 和NO3自由基反应形成[22].从采样点分析,不同利用类型土壤中均检测到 NPAHs.Niederer 等[23]在瑞士巴塞尔游乐园和公园土壤中检测到8 种NPAHs(0.030~0.800µg/kg).Idowu 等[8]检测到澳大利亚工业城市中娱乐区、工业区、吸烟区和居住区土壤中NPAHs 浓度分别为32.7~216.0、98.5~111.0 和 28.6~157.1µg/kg. Garcia-Alonso 等[24]发现西班牙农业、城市和煤气厂土壤中NPAHs 浓度分别为0.184、59.6 和5280µg/kg.De Guidi 等[25]在意大利工业区土壤中检测到3-硝基荧蒽(3-NFlt)、1-硝基芘(1-NPyr)、6-硝基苯并[a]芘(6-NBaP),总浓度在0.053~ 0.260µg/kg.Sun 等[10]调查了我国26 个省份农田土壤中NPAHs,11 种NPAHs 总浓度为(50±45)µg/kg,辽宁、山西、河南和贵州浓度较高.Bandowe 等[16]在青海高原、西安城镇/农村、巢湖盆地以及湛江农业和林业土壤中均检测到 NPAHs(0~10.9µg/kg),4 个气候带中NPAHs 组成存在差异,农村和城市也存在差异.NPAHs 和PAHs 浓度与经度呈正相关,随纬度升高逐渐降低.但此研究只选取了青海、西安、巢湖、湛江4 个城市数据,不具有普遍性.

表2 土壤中硝基多环芳烃污染特征Table 2 The contamination characteristics of NPAHs in soils
从土壤NPAHs 主要成分看,1/2-硝基萘(1/2-NNap, <0.03~205µg/kg)、2-硝基芴(2-NFlu, 0.07~196µg/kg)、9-硝基菲(9-NPhe, 0.011~291µg/kg)、1-NPyr(0.04~1360µg/kg) 、 9- 硝基蒽(9-NAnt,0.011~313µg/kg)、3-NFlt(0.018~3361µg/kg)是土壤中NPAHs 主要成分[8,10,26].Idowu 等[8]发现NPAHs总量与PAHs 呈显著正相关, NPAHs 与PAHs 来源、空间分布可能相同.Cai 等[27]发现土样中1-NNaP 与Nap 呈正相关(r=0.187, P=0.006),但未分析NPAHs总量及其他NPAH.Bandowe 等[16]则证明高原、温带、亚热带土壤中PAHs 总量与NPAHs 呈正相关(r=0.643~0.900, P<0.022),且低分子量和高分子量PAHs 含量均与NPAHs 呈正相关.
2 土壤中ClPAHs 和NPAHs 的迁移转化
土壤既是ClPAHs 和NPAHs 的储存库也是净化场所,土壤中ClPAHs 和NPAHs 可发生吸附-解吸、光降解、氧化还原、生物降解等行为.-Cl 和-NO3使ClPAHs 和NPAHs 的性质和迁移转化规律与母体PAHs 不同.采用EPI Suite 软件评估了ClPAHs 和NPAHs 的物化性质和环境归趋(表3),并总结了它们在土壤中的转化行为及机制(图1).

图1 土壤中ClPAHs 和NPAHs 的转化行为Fig.1 The transformation behaviors of ClPAHs and NPAHs in soils
2.1 土壤中ClPAHs 和NPAHs 的迁移
-Cl 和-NO2为憎水基团,ClPAHs 和NPAHs水溶解度相比PAHs 更低(表3),NPAHs 在土壤中吸附系数和持久性高于母体PAHs[31].Sun 等[10]发现灌溉频繁农业土壤中低水溶性NPAHs 比含氧PAHs有更强固定性、更持久滞留性和更高浓度.PAHs 衍生物在土壤中残留还受有机质、微生物等因素影响,Bandowe 等[16]发现NPAHs 含量与土壤有机碳呈正相关,单独NPAH(1-NNaP、5-NAce、2-NFlu 等)也与有机碳呈正相关.马涛[32]则发现太滆运河流域土壤中总有机碳与高环 PAHs 呈正相关,而与NPAHs 并无显著相关性,因为NPAHs 含量较低.表3显示 PAHs、ClPAHs 和 NPAHs 沸点顺序为NPAHs>ClPAHs>PAHs,土壤中大分子ClPAHs 和NPAHs 比PAHs 更难向空气扩散.而Li 等[6]发现,大气中NPAHs 比PAHs 更易吸附在颗粒物上,容易随颗粒向土壤输入.因此,ClPAHs 和NPAHs 在土壤具有持久性,不易在土壤与水、气界面发生迁移.现在仍无ClPAHs 和NPAHs 在土壤中迁移数据,未来需通过土柱等实验模拟其在土壤中迁移过程.

表3 PAHs、ClPAHs 和NPAHs 物化性质和环境归趋Table 3 The physico-chemical properties and environmental fates of PAHs, ClPAHs, and NPAHs
2.2 土壤中ClPAHs 和NPAHs 的吸附和解吸
ClPAHs 和NPAHs 可在土壤多界面发生吸附-解吸,芳香环上取代基发挥重要作用.以往研究表明,矿物和有机质对芳香烃吸附机制是供体-受体(EDA)作用,包括阳离子-π、n-π、π-π 等[33].-Cl和-NO2是亲电子基,缺电子芳香环和富电子有机质及矿物质(供体)可形成强烈π-π 作用.王珑等[34]提出NPAHs 中芳环上离域电子云π 键受-NO2拉电子作用而缺电子成为电子受体,与黏土矿物中硅氧烷表面氧原子孤对电子n(供体)形成EDA 络合物.-NO2拉电子作用越强,EDA 作用越强烈.而Boyd[35]提出矿物层间水合能力弱的阳离子(K+、NH4+等)可与NPAHs 中-NO2形成Mn+-NO2作用.而-NO2位置影响作用效果,2-硝基萘(2-NNap)和1-硝基萘(1-NNap)中只有2 号位-NO2与萘环共平面,具有较强拉电子作用,与K 基蒙脱石形成KNO2作用[34].NPAHs 分子大小也会影响吸附, 1,5-二硝基萘(163Å2)比1,8-二硝基萘(167Å2)分子面积大,很难进入蒙脱石层间被吸附.其研究仍处于推测阶段,缺少具体数据支持,需采用计算化学方法进一步分析.Al-Bashir 等[36]发现土壤中氨基和硝基Nap吸附行为不同,非电离性硝基Nap 吸附并不随pH值变化,分配系数与溶解度密切相关.1-硝基-2-甲基-萘在土/水中分配系数要高于1-硝基萘,疏水基-CH3降低了1-硝基-2-甲基-萘溶解度.PAHs 衍生物也能被生物体吸附, Choi 等[37]发现短小芽孢杆菌释放的蛋白质对氯代萘有较好吸附能力,疏水作用占据主导,高氯同系物吸附量大于低氯同系物.ClPAHs 在土壤中吸附行为研究匮乏,可借鉴氯苯类污染物研究.Ololade 等[38]发现有机质对氯代苯酚的吸附量与lgKow呈反比, Yang 等[39]则证明矿物是土壤中氯代有机物主要吸附剂,氯代苯酚吸附量与其lgKow呈正比.土壤中ClPAHs 吸附解吸与氯代苯酚有相似之处,但其芳香环种类繁多、取代基复杂,迁移转化规律更加多样.有机质和矿物对污染物吸附规律存在差异,开展研究应针对不同深度和类型土壤具体分析.现有吸附机制研究多限于猜测,未来需通过理论计算结合实验更深入研究.
土壤中ClPAHs 和NPAHs 也会与外来物质发生吸附-解吸行为,影响其环境命运和暴露风险.Zhang等[40]发现聚乙烯等微塑料对9-NAnt 具有高吸附,微塑料疏水表面对疏水有机物具有高亲和性.Hu等[2]的研究同样表明Phe 衍生物在微塑料上的富集依赖于分子疏水性,吸附量与lgKow呈正相关依次为:Phe-Cl > Phe-CH3> Phe > Phe-NO2> PheCHO.分子表面电势显示,-CHO 和-NO2使分子表面电荷更不平衡,导致污染物有弱极性而吸附减弱.微塑料对NPAHs 的吸附受pH 值、盐离子等环境条件影响[40].富电子基-NO2容易与正电荷结合,低pH 下微塑料表面倾向质子化,有助于两者静电吸引[41].盐离子会与NPAHs 竞争位点,微塑料对NPAHs 吸附量随离子浓度增加而下降.Zhang 等[42]指出9-NAnt、微塑料复合暴露中微塑料对9-NAnt 吸附降低了其在生物体内积累,但延长了生物毒性时间,持续对斑马鱼产生神经毒性.而He 等[2]发现吸附有Phe 衍生物的微塑料在胃肠道液中生物可给性在81.34%~98.72%,终身癌症风险高于美国环保局安全限制.微塑料和PAHs 衍生物作为新兴污染物,研究两者在土壤中相互作用及生物毒性具有重要意义,未来仍需重点关注.碳材料作为优良修复材料广泛应用于土壤污染修复,表面积大、官能团丰富的碳材料进入土壤后会与PAHs衍生物发生作用.Li等[43]将活性炭分别加入Pyr 和1-NPyr 污染土壤中,1d 后吸附量分别为78.0%和88.1%.大分子1-NPyr 与活性炭间有更强范德华力,同时1-NPyr 中N 和O 可与活性炭上氢原子形成氢键.而Manousi 等[44]探究了碳材料对2-NFlu、9-NAnt、3-NFlt 等的吸附机理,红外光谱显示苯环C=C 峰向较低波数偏移,N 基团峰也发生明显变化.炭材料中π-电子与NPAHs 中π-电子发生π-π 作用,含氮官能团也参与了相互作用.此外,碳材料对NPAHs 吸附量随其疏水性增加而增加,疏水作用也参与反应.但不少研究提出碳材料有传递电子能力,有毒有机物吸附过程会涉及降解反应,相关研究未考虑到降解反应的可能贡献[45].
2.3 土壤中ClPAHs 和NPAHs 的降解
2.3.1 光降解 PAHs、ClPAHs、NPAHs 光降解是其在土壤中重要消减方式.助色团-Cl 和生色团-NO2使ClPAHs 和NPAHs 吸收光谱发生红移,摩尔吸光系数增加,对日光吸收程度比母体更强[46].NPAHs 光降解半衰期短于母体PAHs, Cvrčková and Ciganek[47]发现Nap、Ant、Chr 在二氯甲烷中光降解半衰期为4252, 8.4, 60d,对应硝基衍生物1-NNap、9-NAnt、6-NChr 半衰期为6, 0.5, 15d.Ohura等[48]发现,ClPAHs 耐光降解能力6-ClBaP<1-ClPy<7-ClBaA ClPAHs 和NPAHs 光降解中间体和产物与母体也具有明显差异. C-C 断裂一般生成含有氯代或硝基小分子有机物,•Cl 或•ON 自由基会与中间体反应生成氯代或硝基有机物.以往研究发现,PAHs在光照下生成有机阳离子自由基,在铁氧化物、黏土矿物等基质表面稳定存在,也叫环境持久性自由基EPFRs[53-54].Zhao 等[52]发现2-ClAnt 和1-ClPyr 在Fe(III)-蒙脱石表面光降解中生成EPFRs,2-ClNap和9-ClPhe 反应体系并未发现.EPFRs 来源于电子传递反应,后两者HOMO 值较低,很难与矿物发生电子传递.Ni 等[55]在1-NNap、9-NAnt、1-NPyr 在赤铁矿光反应体系发现相似规律,1-NNap 光降解中未产生EPFRs.遗憾的是体系未将PAHs 与其衍生物共同研究,未能得到衍生物与母体生成 EPFRs 的差异[52,55].NPAHs 在赤铁矿表面光降解中还可生成•ON 和•NO2活性自由基,CO2存在也可诱导•CO3−生成.PAHs 及其衍生物光修饰和光敏化过程可产生光致毒性,光敏化生成的ROS 会对生物体造成损伤.Onduka 等[56]对10 种NPAHs 在光照和黑暗下急性毒性开展研究,发现除1,5-二硝基萘均具有光致毒性.1-NPyr 光照条件下毒性是黑暗条件下1000多倍,比母体及其羟基和氨基衍生物光致毒性高100 倍,且1-NPyr 在中等光照强度下即可产生光致毒性.鉴于ClPAHs 和NPAHs 的光致毒性效应,未来应重点关注实际土壤中它们的光降解过程和毒性效应. ClPAHs 和NPAHs 光降解会受土壤粒径、厚度等环境因素影响.王依雪等[57]发现2-ClNap 光降解速率随土壤粒径增大而增大,较大土壤颗粒利于通气、透光.土壤颗粒有光屏蔽作用,2-ClNap 光降解速率随土壤厚度增加而减少[53].低pH 值有利土壤中O2•−向H2O2和•OH 转化,促进2-ClNaP 降解.2-ClNap 光降解随腐殖酸含量增高受到抑制.腐殖酸可作为光敏剂诱导1O2、•OH 生成促进光降解,也会与污染物竞争吸收光子产生光屏蔽效应而抑制污染物光降解,说明腐殖酸光屏蔽效应发挥主要作用.Zhao 等[52]发现,环境湿度对ClPAHs 在Fe(III)-蒙脱石光降解具有明显影响,光降解速率随着湿度从10%升高至80%明显受到促进,•OH和O2•−浓度分别增加1.07 倍和62.79 倍.环境因素对ClPAHs 和NPAHs 光降解的影响与母体具有相似性,而其中的差异性仍需要通过大量研究来揭示. 2.3.2 氧化还原降解 有毒有机物被氧化或还原降解能力依赖于自身性质, 亲电子基团-Cl 和-NO2能够显著改变PAHs 氧化还原性质.EPI 评估了污染物被空气氧化半衰期,半衰期顺序为NPAHs>ClPAHs>PAHs,且随取代基团增多而延长(表3).以蒽及其衍生物为例,空气氧化半衰期顺序为9-NO2Ant(2.14d)>9,10-DClAnt(1.30d)>9-ClAnt(0.380d)>Ant(0.267d).而最近研究常采用密度泛函评估污染物分子供/吸电子能力,常用运算符有最高占据分子轨道HOMO、最低未占据分子轨道LUMO、电离能IP、电子亲和度EA等[1,18].IP=7.55eV是微生物和矿物氧化降解 PAHs 阈值,Ant(7.44eV)、Pyr(7.43eV)、BaP(7.10eV)会被锰过氧化物酶和含铁黏土矿物氧化降解[58].HOMO 值可用于评估分子给电子能力,本文采用密度泛函理论计算了常见ClPAHs、NPAHs和PAHs 的HOMO 值.图2 显示HOMO 值顺序为NPAHs 图2 PAHs、ClPAHs 和NPAHs 的HOMO 值和轨道分布Fig.2 The HOMO values and orbital distribution of PAHs, ClPAHs, and NPAHs 2.3.3 生物降解 微生物是土壤中PAHs 及其衍生物转化主要驱动力,取代基种类、数量、位置等会影响降解菌种、速率和途径[64].表3 中快速生物降解性顺序为PAHs> ClPAHs > NPAHs,随着取代基增多而减小.Heitkamp 等[65]将石油污染沉积物中分离的PAHs 降解微生物Mycobacterium sp.用于1-NPyr 降解,培养10d 后矿化率仅有12.3%.好氧和厌氧体系中降解均缓慢,好氧体系检测到少量14CO2,厌氧体系则没有14CO2生成,1-NPyr 被还原为1-氨基芘.PAHs 高降解菌很难降解NPAHs,硝基引入改变了代谢途径并减缓降解速率.而Walker 等[66]分离的萘降解土著菌可将1-ClNap 降解生成萘羟基衍生物和水杨酸衍生物. Mori 等[67]发现白腐真菌Phlebia lindtneri 可将1/2-ClNap 加氧生成羟基氯代衍生物,但不能实现脱氯和苯环断裂,P450 单加氧化酶发挥主要作用.而马涛等[68]则发现黄孢原毛平革菌和9,10-DClAnt 反应过程中则发生脱氯反应和开环反应.除P450 单加氧酶外,木质素过氧化物酶、锰过氧化物酶等也发生作用.过氧化物酶利用H2O2使氯蒽发生单电子氧化,脱氯生成9,10-蒽醌,进而开环生成酮、酸、酯类化合物[69-70]. NPAHs 降解机理与PAHs 不同,与硝基芳烃相似,可作为碳源和氮源被微生物降解[70].Li 等[71]从富含1-NNap 的美国新泽西州化工土壤中分离出Sphingobium sp.菌株JS3065,发现编码1-NNap 代谢基因簇位于质粒上,与编码萘分解基因簇具有共同起源.分解起始,双加氧酶作用下1-NNap 生成1,2-羟基萘,随后降解途径与Nap 一致.好氧条件通过氧化和还原途径生物降解NPAHs,厌氧体系通过还原途径降解[71].RAFII 等[72]发现Mycobacterium sp.也可通过氧化和还原途径降解1,3-DNPyr、1,6-DNFlu和6-NChr 等,还原过程产物为氨基PAHs. Pothuluri等[73]发现真菌Cunninghamella elegans 可通过P450单加氧酶和环氧化物水解酶将1-NPyr、6-NBaP、2/3-NFlt、2-NFlu 和1-NChr 氧化为硝基芳烃氧化物和硝基芳烃反式二氢二醇,随后发生重排并与硫酸盐、葡萄糖等偶联生成硝基硫酸酯而实现解毒.Al-Bashir 等[36]发现土壤中硝基萘矿化第一阶段由微生物控制快速降解,动力学曲线符合酶催化反应Michaelis-Menten 模型.第二阶段降解缓慢,土壤上污染物未饱和,生物降解受其解吸速率影响.同时发现硝基萘矿化速率是氨基萘2~4 倍,但矿化时间滞后4~6 周.研究者并未解释相关机制,结合后期NPAHs降解途径研究猜测,硝基萘先被还原酶快速还原成羟胺,随后同氨基萘一样缓慢矿化.1-硝基-2-甲基-萘降解速率要慢于1-硝基萘,-CH3基团会减少加氧酶对萘环攻击位点数.研究发现,碳材料可增强NPAHs生物降解.Wang 等[74]将炭黑加入希瓦氏菌MR-1 与硝基联苯体系中,污染物还原速率常数从0.0044h−1提高到0.035h−1.增强倍数与炭黑电导率呈正相关,炭黑在污染物生物还原过程可能充当了导体作用.ClPAHs 和NPAHs 生物降解文献报道比较陈旧,应结合土壤污染特征,开展主要污染物降解菌筛选,加强降解途径和降解产物毒性效应研究. ClPAHs 和NPAHs 浓度低于母体PAHs(0~5)数量级,ClPAHs 集中在典型污染场地和附近土壤,NPAHs 在各种土壤中广泛存在.土壤中ClPAHs和NPAHs 迁移转化与母体PAHs 有明显区别,易发生光降解,难以迁移且不易被矿物和微生物转化.现有研究仍存在不足,建议从以下几个方面进行深入研究. 3.1 国内外还没有PAHs 衍生物标准检测方法,且土壤中污染数据仍然较少.受检测技术限制, PAHs衍生物检测准确度、精度以及种类很难满足需求.未来需更多PAHs 衍生物标准品商业化,实现土壤中更多PAHs 衍生物精准定性和定量.优化前处理技术,实现PAHs 衍生物快速、简便、完全提取.只有将这些技术发展完善,才能准确掌握土壤中PAHs 衍生物赋存形态和浓度.未来还需系统研究各区域、利用方式土壤中PAHs 衍生物污染水平,为土壤PAHs 衍生物污染现状和管控研究提供基础数据. 3.2 PAHs衍生物物化特性多依赖于EPI Suite软件和模型数据,仍缺少实际实验数据.PAHs衍生物在土壤中垂直分布、土壤颗粒和孔隙水间分配行为尚未见报道,需进行土柱实验模拟其在土壤中垂直迁移.除Nap 衍生物外,土壤组分对PAHs 衍生物吸附能力和吸附机理尚不清晰.未来需通过实验方法结合理论计算,更全面、深入地掌握PAHs 衍生物在土壤多界面的迁移过程. 3.3 PAHs 衍生物在土壤及组分界面降解研究仍然匮乏.降解数据以萘衍生物为主,其他PAH 的衍生物降解研究相对较少.受PAHs 疏水性影响,PAHs 转化研究多集中于干燥土壤矿物,未来可关注土壤活性自由基介导下水相中PAHs 衍生物转化机制.土壤组分和环境条件对降解的影响规律仍不清晰,未来需关注Cl 源和N 源存在下ClPAHs 和NPAHs 转化和形成机制.Cl/N 同位素标记、傅里叶变换回旋共振质谱等技术对探究PAHs 转化机制也有重要帮助.
3 结论和展望
猜你喜欢
辽河(2021年10期)2021-11-12化工管理(2021年7期)2021-05-13中国环境科学(2019年7期)2019-07-31中国农资(2016年1期)2016-12-01化工进展(2015年3期)2015-11-11中国当代医药(2015年20期)2015-03-01火炸药学报(2014年1期)2014-03-20无机化学学报(2014年8期)2014-02-28当代修辞学(2014年3期)2014-01-21中国科学技术大学学报(2013年8期)2013-03-11
