
通过密度泛函理论对溶剂效应对碳氢键断裂步骤的影响研究
2024-06-16 05:41田腾安会勇王涛于芳
辽宁化工 2024年5期
田腾 安会勇 王涛 于芳
摘 要: 碳氢键活化反应在合成方面的作用已经被研究证明了。其中8-氨基喹啉导向基介导的碳氢键活化反应是实现此策略最常用的方法。大量的计算化学研究都已证明8-氨基喹啉导向基自身强大的供电子效应以及刚性结构是其反应高效的关键。然而,与此相关的溶剂效应并没有得到充分研究。依次选取极性从低到高的溶剂甲苯、1,2-二氯乙烷、丙酮、乙腈、二甲基亚砜的隐式溶剂模型来对8-氨基喹啉导向基介导的sp3碳氢键活化反应的决速步(碳氢键断裂步骤)进行密度泛函理论(DFT)计算,最终得知随着溶剂极性的提高,碳氢键断裂步骤的过渡态的熵减程度会明显下降,从而使得对应的吉布斯自由能垒降低,进而加快碳氢键断裂步骤的进行。
关 键 词:碳氢键活化;密度泛函理论;吉布斯自由能
中图分类号:TQ013.1 文献标识码: A 文章编号: 1004-0935(2024)05-0667-04
碳氢键活化作为一种有效的合成策略,已被广泛研究了近20年之久[1]。为了实现这一策略,多种碳氢键活化方法被陆续地开发了出来,其中被研究和应用最广泛的是导向基介入的碳氢键活化策略。此方法近年来也取得了较大的突破,以最原始的双齿配体导向基[2-7]为起点,结构上更加简单的单齿配体导向基[8-14]、可以活化更加远位置的远程导向 基[15-17]、可在反应中自动安装拆解的瞬时导向 基[18-21]等,都被逐渐研究了出来,并在各种体系中展现了其功能强大之处。但是由于碳氢键的键能比较高,并且相对来说比较稳定,如果对其直接进行官能团化,那么毫无疑问是十分困难的。另外,一个有机化合物分子内通常有很多种性质不同的碳氢键,如何对其某一种特定的碳氢键进行活化,而不影响分子中其他种类的碳氢键的性质,这就是碳氢键活化过程中的选择性问题。碳氢键活化反应,就是在某种特定的条件下,对某种有机化合物中的某种特定类型的碳氢键进行活化,使其反应性增强或切断,从而实现定向的化学转化。因此,科研工作者们所面临的最大问题便是如何对碳氢键进行活化,以及如何解决其进行化学反应时的选择性问题。如果能够选择性切断碳氢键,并开发出实用的合成化学新反应,毫无疑问,必将会推动整个有机行业的发展。
虽然导向基介入的碳氢键活化策略中包含了很多的方法,但是目前来说,应用最为广泛的还是 8-氨基喹啉导向基(AQ)[2]。原因是这种导向基所能完成的碳氢键活化反应种类是最多的,如芳基 化[22-28]、氨基化[29]、醚化[30-31]、酯基化[32-33]等,除此之外,其还可以在相对廉价的第一过渡系金属催化下[34-35],相对高效地完成各种碳氢键活化反应,这是绝大多数导向基所无法完成的。为了解释这种导向基的强大性能[36-37],大量的计算化学研究工作层出不穷。如利用密度泛函理论(DFT),对其在反应体系中的过渡态能垒进行计算,以及利用波函数分析法对其在反应中的电子效应、位阻效应、轨道效应等进行了细致的分析,得出的结论是它们强大的供电子性能以及相对刚性的分子结构,使得过渡金属能够比其他在使用导向基团的时候更加平稳地完成目标反应。然而,在这些研究中,溶剂效应并没有被细致地研究。
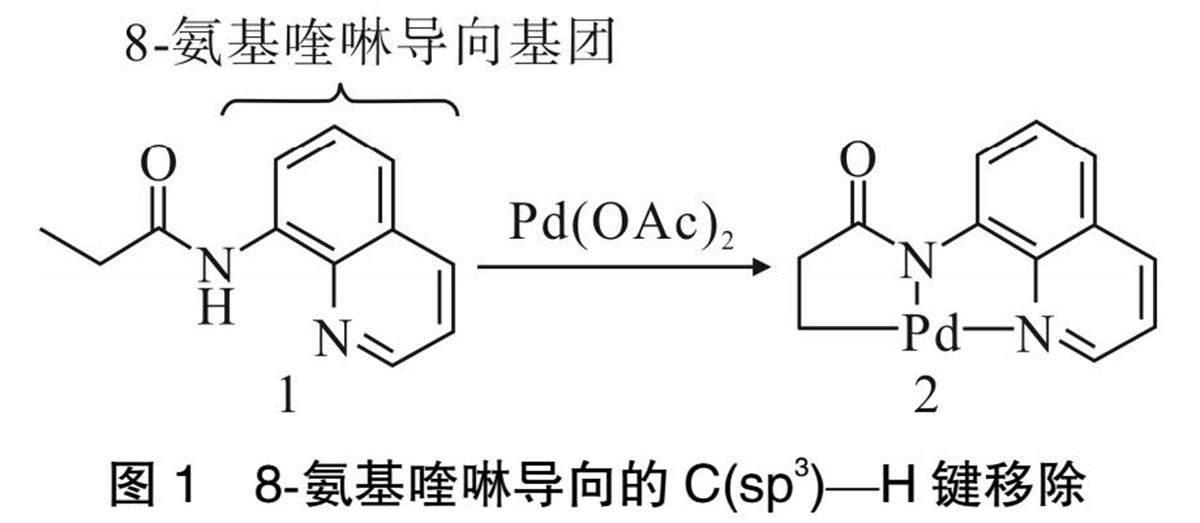
本文将对AQ导向基参与的sp3碳氢键活化反应中的碳氢键断裂步骤(如图1所示)进行DFT理论计算[42-43],通过计算不同的隐式溶剂模型下的反应能垒数据,分析不同种类的溶剂对碳氢键活化反应的影响,从而对后续的实验研究提供相对应的理论依据。
1 实验部分
1.1 计算硬件
服务器配置(Linux):E5-2696 v4*2,双路 44核/88线程,128 GB内存。GPU加速服务器(Linux):E5-2678 v3*2,双路24核/48线程,64 GB内存。
1.2 实验方法
使用量子化学计算软件-Gaussian(版本Gaussian 16, Revision A.03),对AQ导向基、参与的sp3碳氢键活化反应中的碳氢键断裂(sp3碳氢键活化反应中,通常为决速步骤)进行DFT理论计算,通过计算不同隐式溶剂模型的反应体系下(甲苯、1,2-二氯乙烷,丙酮、乙腈、二甲基亚砜)的反应能垒数据,分析不同介电常数的溶剂对碳氢键活化反应的影响。所有几何优化和频率计算均使用色散校正混合GGA函数PBE0-D3进行计算,Pd在计算时使用def2tzvp基组、其他原子则使用def2svp基组。通过内禀反应坐标(IRC)计算,将所有定位的过渡态连接到最近的最小值,并在相同的理论水平上计算,随后对所得结构进行优化。对优化得到的几何结构进行了振动分析,以通过分别存在0或1个虚频来验证它们是最小点或鞍点。使用SMD连续溶剂化模型将甲苯、1,2-二氯乙烷、丙酮、乙腈、二甲基亚砜的溶剂效应纳入计算。在def2tzvpp基组下计算体系的单点能量。在383.15 K的反应温度下计算焓和吉布斯自由能。
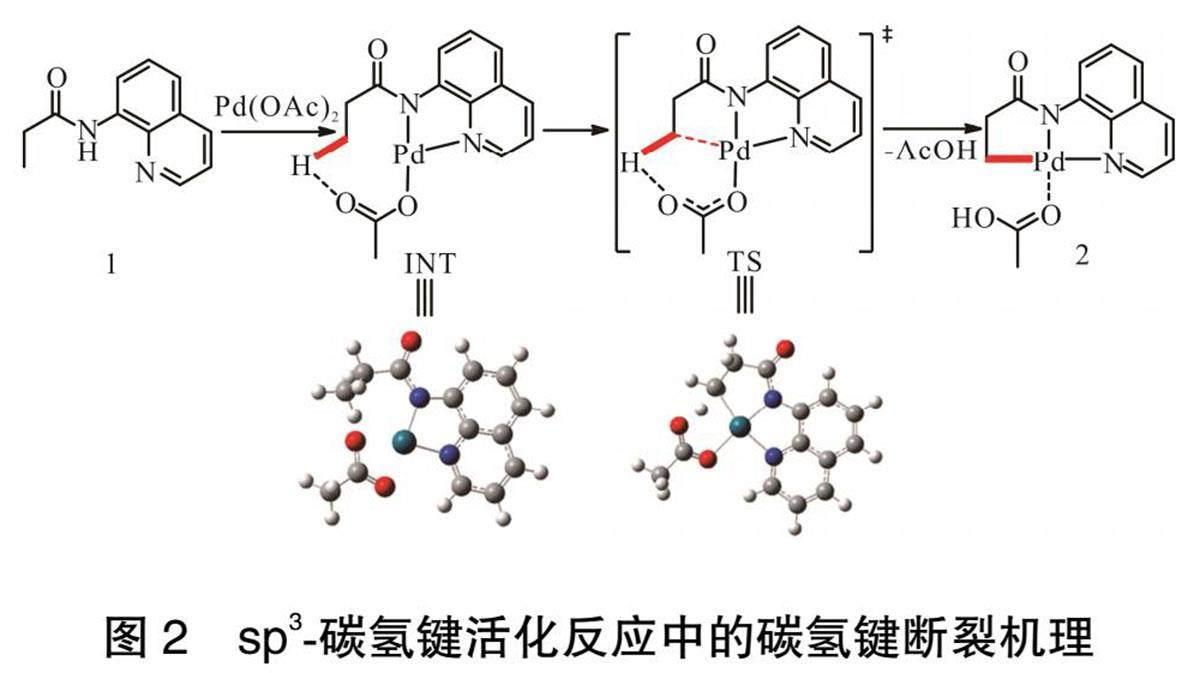
碳氢键断裂机理如图2所示。导向基团1与催化剂醋酸钯通过1个氧氢之间的氢键作用螯合形成环钯中间体INT,之后C(sp3)与钯进行作用,随同醋酸根和C(sp3)—H键形成过渡态TS,最后在一步协同过程中,醋酸根拔取C(sp3)—H键上的H,并且C(sp3)与Pd之间形成碳钯键结构2。
2 结果与讨论
2.1 不同种类的溶剂对于碳氢键断裂过渡态焓垒与熵垒的影响
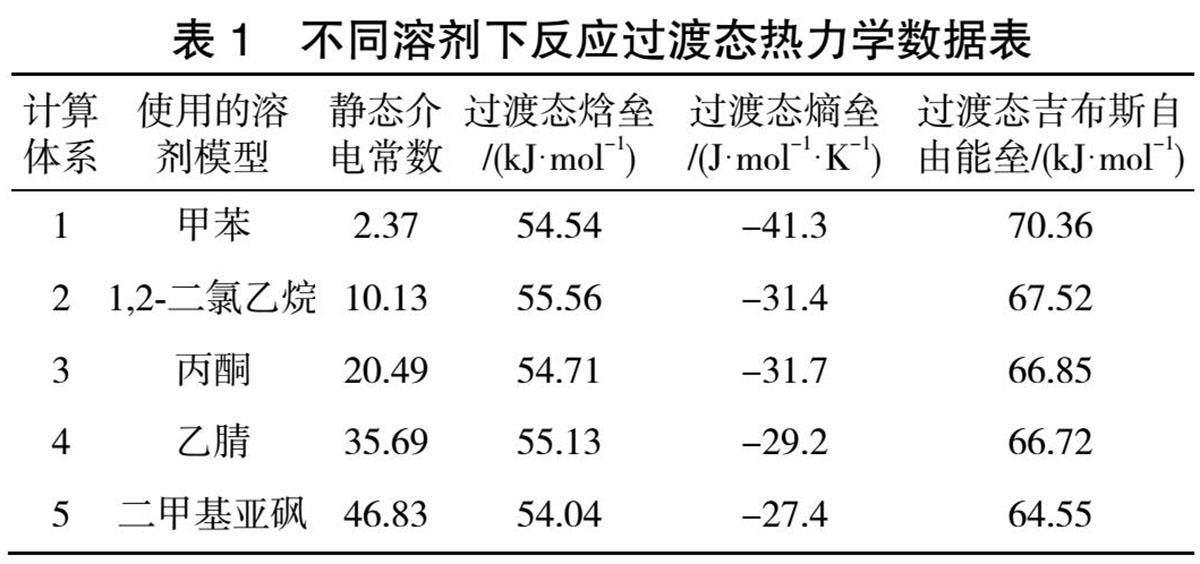
不同的溶剂体系中,其对应的溶剂参数不同,其中影响反应最大的参数就是静态介电常数。因此,挑选了一系列静态介电常数从大到小的溶剂(极性从小到大的一系列溶剂),即甲苯、1,2-二氯乙烷、丙酮、乙腈、二甲基亚砜,来探究不同极性溶剂对于碳氢键断裂过渡态动力学数据上的影响,以得知反应的难易程度。
首先,先对过渡态焓垒和熵垒进行了计算与分析,结果如表1所示。由表1可知,随着所使用的溶剂的极性逐步增大,不同体系之间的焓垒没有十分明显的差别,都在54.42 kJ·mol-1左右,说明溶剂的极性没有对碳氢键断裂这一步的断键与成键的难易程度造成很大的影响,也暗示了溶剂分子在反应中无法到反应核心区,因此也无法利用其极性区的电子云与反应分子的亲电部分结合来影响反应的主要的断键成键过程。
反观不同溶剂体系下的熵垒差别尤为明显。从整体来看,此步骤是熵减反应,同时随着使用溶剂的极性增大,过渡态的焓下降得越少,即混乱度越大。 这说明在高极性溶剂体系下进行该反应,需要反应物所克服的外部溶剂的包裹的难度越大,也间接说明了在该反应中溶剂只是在反应体系外部进行包裹,并没有深入反应核心。
2.2 不同种类的溶剂对于碳氢键断裂过渡态吉布斯自由能垒的影响
仅对过渡态的焓垒和熵垒进行分析还无法全面地得知碳氢键断裂在不同溶剂下发生反应的难易程度,因此对吉布斯自由能垒也进行了计算与分析,结果如表2所示。
由表2可以看出,吉布斯自由能垒随着溶剂极性的增大而相对明显地逐步降低,也就是说更加容易发生碳氢键断裂反应。
综合焓垒、熵垒、吉布斯自由能垒来看,虽然从吉布斯自由能垒数据中直接得出了该反应在高极性溶剂中更容易发生,但是实际上吉布斯自由能是由焓、熵、温度计算而来,因此只从吉布斯自由能垒数据得出结论是不够的,在之前的讨论中熵减程度随着溶剂极性的增大而显著减小,焓垒则基本没有什么变化,因此可以得出最终结论:使用的溶剂极性越大,会使得过渡态的熵减程度减小,从而使得对应的吉布斯自由能垒变小,从而加快碳氢键断裂反应。
3 结 论
8-氨基喹啉(AQ)导向基介导的碳氢键活化反应一直在被广泛研究,本工作对其在sp3碳氢键活化中的决速步即碳氢键断裂步骤在不同的溶剂模型下,用密度泛函理论(DFT)进行了过渡态能垒的计算。计算结果显示,随着溶剂极性的提高,其过渡态的熵减程度会明显下降,从而使得对应的吉布斯自由能垒降低,加快碳氢键断裂步骤的进行。本工作将为之后的碳氢键活化的计算和实验工作提供参考。
参考文献:
[1] LIU B, ROMINE A M, RUBEL C Z, et al. Transition-metal-catalyzed, coordination-assisted functionalization of nonactivated C(sp3)–H bonds[J]. Chemical Reviews, 2021, 121(24): 14957-15074.
[2] SHABASHOV D, DAUGULIS O. Catalytic coupling of C?H and C?I bonds using pyridine as a directing group[J]. Organic Letters, 2005, 7(17): 3657-3659.
[3] ZHANG Q, CHEN K, RAO W, et al. Stereoselective synthesis of chiral α-amino-β-lactams through Palladium(II)-catalyzed sequential monoarylation/amidation of C (sp3)–H bonds[J]. Angewandte Chemie, 2013, 125(51): 13833-13837.
[4] AFFRON D P, BULL J A. Palladium-catalyzed directed C (sp3)–H arylation of saturated heterocycles at C-3 using a concise optimization approach[J]. European Journal of Organic Chemistry, 2016, 2016(1): 139-149.
[5] ZHANG G, XIE X, ZHU J, et al. Pd (II)-catalyzed C(sp3)–H arylation of amino acid derivatives with click-triazoles as removable directing groups[J].Organic & Biomolecular Chemistry, 2015, 13(19): 5444-5449.
[6] HAN J,ZHENG Y,WANG C,et al. Palladium-catalyzed oxalyl amide-directed γ-arylation of aliphatic amines[J]. The Journal of Organic Chemistry, 2015, 80(18): 9297-9306.
[7] HAN J, ZHENG Y, WANG C, et al. Pd-catalyzed coupling of γ-C (sp3)–H bonds of oxalyl amide-protected amino acids with heteroaryl and aryl iodides[J]. The Journal of Organic Chemistry, 2016, 81(13): 5681-5689.
[8] LE K K A, NGUYEN H, DAUGULIS O. 1-Aminopyridinium ylides as monodentate directing groups for sp3 C–H bond functionalization[J]. Journal of the American Chemical Society, 2019, 141(37): 14728-14735.
[9] PENG J, CHEN C, XI C. β-Arylation of oxime ethers using diaryliodonium salts through activation of inert C (sp3)–H bonds using a palladium catalyst[J]. Chemical science, 2016, 7(2): 1383-1387.
[10] STOWERS K J, FORTNER K C, SANFORD M S. Aerobic Pd-catalyzed sp3 C?H olefination: a route to both N-heterocyclic scaffolds and alkenes[J]. Journal of the American Chemical Society, 2011, 133(17): 6541-6544.
[11] YANG W, YE S, SCHMIDT Y, et al. Ligand-promoted C(sp3)?H olefination en route to multi-functionalized pyrazoles[J]. Chemistry–A European Journal, 2016, 22(21): 7059-7062.
[12] HE C, GAUNT M J. Ligand-assisted palladium-catalyzed C–H alkenylation of aliphatic amines for the synthesis of functionalized pyrrolidines[J]. Chemical Science, 2017, 8(5): 3586-3592.
[13] WASA M, ENGLE K M, YU J Q. Pd (II)-catalyzed olefination of sp3 C?H bonds[J]. Journal of the American Chemical Society, 2010, 132(11): 3680-3681.
[14] LI S, CHEN G, FENG C G, et al. Ligand-enabled γ-C–H olefination and carbonylation: construction of β-quaternary carbon centers[J]. Journal of the American Chemical Society, 2014, 136(14): 5267-5270.
[15] LIU X, SONG Y, LIU A, et al. More than a leaving group: N-phenyltrifluoroacetimidate as a remote directing group for highly α-selective 1,2-cis glycosylation[J]. Angewandte Chemie, 2022, 134 (21): e202201510.
[16] HETTIKANKANAMALAGE A A, LASSFOLK R, EKHOLM F S, et al. Mechanisms of stereodirecting participation and ester migration from near and far in glycosylation and related reactions[J]. Chemical reviews, 2020, 120(15): 7104-7151.
[17] YANG B, YANG W, RAMADAN S, et al. Pre-activation-based stereoselective glycosylations[J]. European journal of organic chemistry, 2018, 2018(9): 1075-1096.
[18] LI Y H, OUYANG Y, CHEKSHIN N, et al. PdII-Catalyzed site-selective β-and γ-C(sp3)–H arylation of primary aldehydes controlled by transient directing groups[J]. Journal of the American Chemical Society, 2022, 144(11): 4727-4733.
[19] ZHANG F L, HONG K, LI T J, et al. Functionalization of C(sp3)–H bonds using a transient directing group[J]. Science, 2016, 351(6270): 252-256.
[20] WANG P, VERMA P, XIA G, et al. Ligand-accelerated non-directed C–H functionalization of arenes[J]. Nature, 2017, 551(7681): 489-493.
[21] XU Y, YOUNG M C, WANG C, et al. Catalytic C (sp3)?H arylation of free primary amines with an exo directing group generated in situ[J]. Angewandte Chemie International Edition, 2016, 55(31): 9084-9087.
[22] HILL D E, YU J Q, BLACKMOND D G. Insights into the role of transient chiral mediators and pyridone ligands in asymmetric Pd-catalyzed C–H functionalization[J]. The Journal of Organic Chemistry, 2020, 85(21): 13674-13679.
[23] LIU L Y, QIAO J X, YEUNG K S, et al. Meta-selective C? H arylation of fluoroarenes and simple arenes[J]. Angewandte Chemie, 2020, 132(33): 13935-13939.
[24] SHI H, LU Y, WENG J, et al. Differentiation and functionalization of remote C–H bonds in adjacent positions[J]. Nature chemistry, 2020, 12(4): 399-404.
[25] HU L, MENG G, YU J Q. Ligand-enabled Pd (II)-catalyzed β-methylene C (sp3)–H arylation of free aliphatic acids[J]. Journal of the American Chemical Society, 2022, 144(45): 20550-20553.
[26] LI Z, PARK H S, QIAO J X, et al. Ligand-enabled C–H hydroxylation with aqueous H2O2 at room temperature[J]. Journal of the American Chemical Society, 2022, 144(39): 18109-18116.
[27] LI Y H, OUYANG Y, CHEKSHIN N, et al. PdII-Catalyzed γ-C (sp3)–H (Hetero) arylation of ketones enabled by transient directing groups[J]. ACS Catalysis, 2022, 12(17): 10581-10586.
[28] FAN Z, CHEN X, TANAKA K, et al. Molecular editing of aza-arene C–H bonds by distance, geometry and chirality[J]. Nature, 2022, 610(7930): 87-93.
[29] OLAH G A, ERNST T D. Trimethylsilyl azide/triflic acid, a highly efficient electrophilic aromatic amination reagent[J]. The Journal of Organic Chemistry, 1989, 54(5): 1203-1204.
[30] VOUGIOUKALAKIS G C, CHRONAKIS N, ORFANOPOULOS M. Addition of electron-rich aromatics to azafullerenium carbocation. a stepwise electrophilic substitution mechanism[J]. Organic Letters, 2003, 5(24): 4603-4606.
[31] WANG J, ZHOU Y, XU X, et al. Entry to 1,2,3,4-tetrasubstituted arenes through addressing the “meta constraint” in the palladium/norbornene catalysis[J]. Journal of the American Chemical Society, 2020, 142(6): 3050-3059.
[32] KIM J, JOO J M. Palladium-catalyzed C–H acetoxylation of arenes using a pyrazolonaphthyridine ligand[J]. Bulletin of the Korean Chemical Society, 2022, 43(10): 1173-1176.
[33] MUKHOPADHYAY S, ROTHENBERG G, LANDO G, et al. Air oxidation of benzene to biphenyl–a dual catalytic approach[J]. Advanced Synthesis & Catalysis, 2001, 343(5): 455-459.
[34] 步亚楠.铁催化的N-烷基化反应研究进展[J].辽宁化工,2022,51(8):1118-1120.
[35] PHIPPS R J, GRIMSTER N P, GAUNT M J. Cu (II)-catalyzed direct and site-selective arylation of indoles under mild conditions[J]. Journal of the American Chemical Society, 2008, 130(26): 8172-8174.
[36] 潘永凯,陈运荣. 手性噁唑啉的不对称催化合成研究进展[J]. 辽宁石油化工大学学报,2021,41(4):9-16.
[37] 朱超凡,王锐,陈运荣. 2-乙基-2-芳基-二氢喹啉衍生物的合成[J].辽宁石油化工大学学报,2022,42(5):18-25.
Study of Solvent Effect on C—H Cleavage Step
by Density Functional Theory
TIAN Teng1,2, AN Huiyong1*, WANG Tao3*, YU Fang1,2*
(1. School of Petrochemical Engineering, Liaoning Petrochemical University, Fushun Liaoning 113001, China;
2. Ningbo Institute of Dalian University of Technology, Ningbo Zhejiang 315016, China;
3.Wuxi Institute of Drug Control, Wuxi Jiangsu 214000, China)
Abstract: The efficiency of C—H activation has been widely studied and proved. 8-aminoquinoline mediated C—H activation is the most commonly utilized method. A large number of computational chemistry studies have proved that the powerful electron-donating effect and rigid structure of 8- aminoquinoline guiding group are the key to its efficient reaction. However, the solvent effect related to this system has not been fully interpreted. In this paper, the continuum solvation models of toluene, 1,2-dichloroethane, acetone, acetonitrile and dimethyl sulfoxide were selected in order to engage the density functional theory (DFT) calculation of the RDS of the sp3C—H activation mediated by 8-aminoquinoline guiding group (C—H cleavage step). Finally, it was found that with the increase of solvent polarity, the entropy reduction of the transition state of the C—H cleavage step decreased significantly that caused the reduction of Gibbs free energy barrier, which finally accelerated the reaction.
Key words: C—H activation; Density functional theory; Gibbs free energy
基金项目:辽宁省重点研发计划项目(项目编号:2019JH2/10100005)。
收稿日期:2023-03-13
作者简介:田腾(1997-),男,陕西省渭南市人,硕士研究生,研究方向:过渡金属催化碳氢键活化反应。
通信作者:安会勇(1980-),男,教授,博士,研究方向:有机合成方面研究。
王涛(1988-),男,硕士,主管药师,研究方向:药物检测与分析。
于芳(1984-),女,博士,副教授,研究方向:有机凝胶合成研究。
猜你喜欢
燃料化学学报(2023年3期)2023-03-11
北京航空航天大学学报(2022年5期)2022-06-06
大学化学(2021年8期)2021-09-26
中学课程辅导·教学研究(2021年8期)2021-07-14
燃料化学学报(2021年5期)2021-06-02
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
大学化学(2016年4期)2016-07-27
学园(2015年5期)2015-10-21
中国有色金属学报(2011年4期)2011-11-08
