
丙硫菌唑原药中3 个主要杂质合成及产生历程分析
2024-03-19 11:25杨丙连王建博丁亚伟
世界农药 2024年2期
杨丙连,王建博,丁亚伟
(1.南通泰禾化工股份有限公司,江苏 南通 226407;2.上海泰禾化工有限公司,上海 201612)
丙硫菌唑(prothioconazole,CAS:178928-70-6)作为全球第二大杀菌剂,是拜耳作物科学公司成功开发的新型三唑硫酮类高效广谱杀菌剂,作用机制为抑制真菌甾醇合成,可用于谷物、大豆和棉花等作物多种病害的防治[1-3]。
丙硫菌唑的主流合成工艺是以邻氯氯苄为原料,与镁反应生成格氏试剂,再与2-氯-1-(1-氯环丙基)乙酮发生加成反应,然后经肼化、关环、氧化等一系列反应制备得到丙硫菌唑[4-5],具体合成路线如图1。
基于原药分析和文献调研,发现丙硫菌唑合成过程会产生图2 所示的3 个主要杂质:2-[2-(1-氯环丙基)-2-羟基-3-苯基丙基]-2,4-二氢-3H-1,2,4-三唑-3-硫酮(简称脱氯丙硫,DC-PTC)、2-[2-(1-氯环丙基)-1-(2-氯苯基)-3-(1H-1,2,4-三唑-1-基)丙烷基]-2-醇(简称脱硫丙硫,DS-PTC)和2-{2-(1-氯环丙基)-1-(2-氯苯基)-3[5-(甲巯基)-1H-1,2,4-三唑-1-基]丙烷基}-2-醇(简称甲基化丙硫,Me-PTC)。

图2 丙硫菌唑原药中3 个主要杂质
杂质不仅影响原药品质,且杂质的毒理学特性与丙硫菌唑差异较大,在原药中均有明确限量要求。如美国环境保护署2007 年发布的丙硫菌唑人类健康风险评估报告中表明杂质DS-PTC 具有致畸性,会导致幼儿畸形,其在原药中的含量要求低于500 mg/L。
据文献调查,尚无丙硫菌唑上述3 个杂质产生原因的分析和合成方法报道。本文基于丙硫菌唑原药合成路线分析推测了杂质产生的可能历程,并设计了下述合成路线对3 个杂质进行了合成,其结构经LC-MS、NMR 等表征确证,为丙硫菌唑工艺优化、杂质和产品质量控制提供参考。
脱氯丙硫杂质(DC-PTC)合成路线:
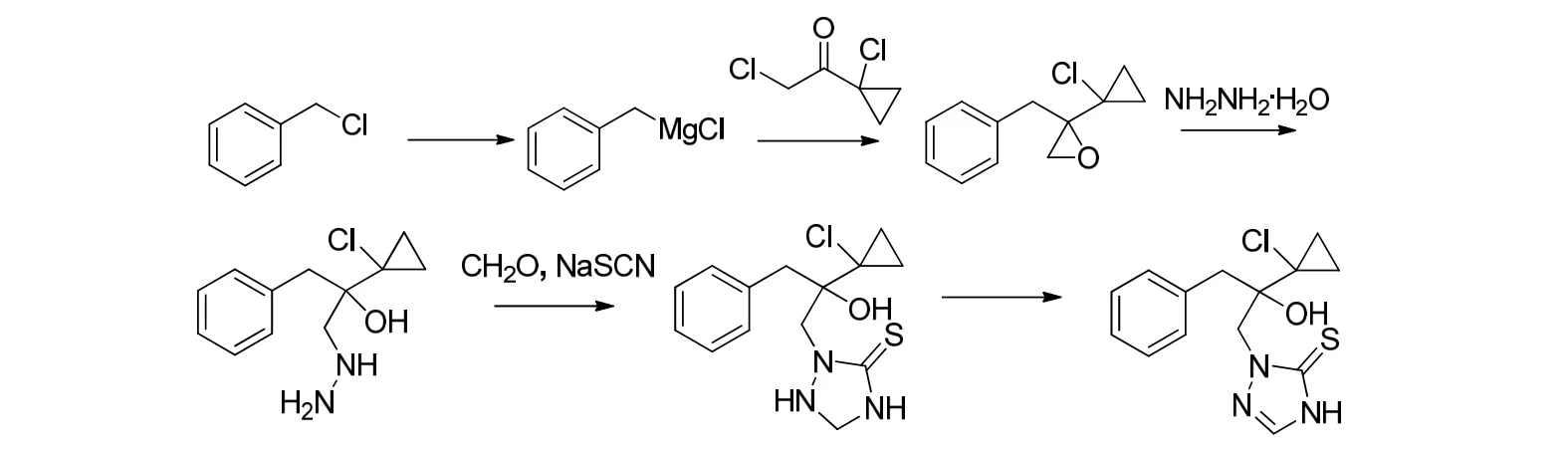
脱硫丙硫杂质(DS-PTC)合成路线:
甲基化丙硫杂质(Me-PTC)合成路线:
1 实验部分
1.1 试剂和仪器
试剂:甲醇、甲苯、水合肼、硫氰酸钠、碘甲烷、无水碳酸钾、氯化苄、镁屑、氢氧化钠和1,2,4-三氮唑等均为分析纯,均购自国药试剂;2-氯-1-(1-氯环丙基)乙酮(90%)和2-(1-氯环丙基)-2-(2-氯苯基)甲基环氧乙烷(75%)为实验室自制。
仪器:多电子天平(梅特勒-托利公司)、GC-14C岛津气相色谱仪(日本岛津公司)、DionexUltimate TM 3000 超高效液相色谱仪(美国Dionex 公司);智能数显恒温油/水浴锅和机械搅拌器(巩义市予华仪器有限责任公司);AV400 型(400 Hz)核磁共振仪(瑞士Bruker 公司);Q-ExactiveTM 组合型四极杆-高分辨轨道阱质谱仪[赛默飞世尔科技(中国)有限公司]。
1.2 杂质的合成
1.2.1 2-[2-(1-氯环丙基)-2-羟基-3-苯基丙基]-2,4-二氢-3H-1,2,4-三唑-3-硫酮(DC-PTC)的合成
2-(1-氯-环丙基)-2-(苯基)甲基环氧乙烷:500 mL四口反应瓶中加入镁屑(7.3 g,0.3 mol),甲苯(15.0 g)与四氢呋喃(15.0 g)的混合溶剂30.0 g,氮气置换3 次,开启搅拌,升温至35~40 ℃,滴加氯化苄(25.3 g,99%,0.2 mol)与甲苯(50.0 g)、四氢呋喃(50.0 g)的混合液,滴完后保温15 min,氮气保护下将上清液转移至另一500 mL 反应瓶中。开启搅拌,降温至10~15 ℃,于30 min左右滴加29.0 g(95.0%,0.18 mol)2-氯-1-(1-氯环丙基)乙酮,继续保温反应20 min,得到浅黄色透明格氏加成液。将上述加成液倒入200 g冰水中淬灭,用浓盐酸调pH=5~6,充分搅拌约30 min后静置分层,有机相依次用水、饱和食盐水洗涤,再用无水硫酸钠干燥,过滤,脱除溶剂得41.8 g(69.9%,0.14 mol)2-(1-氯-环丙基)-2-(苯基)甲基环氧乙烷粗品,不经纯化直接投下一步反应。
2-(1-氯-环丙-1-基)-3-(苯基)-2-羟基-丙基-1-肼盐酸盐:于500 mL 四口瓶中依次投入甲醇(100 g)、水合肼(44.0 g,80%,0.70 mol)、氢氧化钠(6.2 g,96%,0.15 mol)和2-(1-氯环丙基)-2-(苯基)甲基环氧乙烷粗品(41.8 g,69.9%,0.14 mol),氮气保护下升温至回流反应2 h。将上述反应液脱除甲醇,残余物用甲苯(100 g)充分溶解后分层,有机相经水洗涤2 次。再向该有机相滴入浓盐酸(16.6 g,37%,0.17 mol),控制内温不超过35 ℃,滴完后搅拌2 h 至固体完全析出,过滤,干燥后得到32.0 g 浅黄色粉末状2-(1-氯环丙基)-3-(苯基)-2-羟基-丙基-1-肼盐酸盐(含量97.1%)。
2-(1-氯-环丙-1-基)-1-(苯基)-2-羟基-3-(1,2,4-三唑烷-5-硫羰-1-基)-丙烷:于500 mL 四口瓶中加入甲苯和上一步骤得到的2-(1-氯环丙基)-3-(苯基)-2-羟基-丙基-1-肼盐酸盐(32.0 g,97%,0.11 mol),氮气保护下滴入氢氧化钠溶液(17.6 g,30%,0.13 mol)进行反应,约30 min 后加入甲醛溶液(8.9 g,37%,0.11 mol)和硫氰酸钠(9.1 g,99%,0.11 mol),搅拌反应约30 min 后滴入盐酸(12.8 g,37%,0.13 mol),全程控制反应温度20~30 ℃,保温反应约4 h。过滤,滤饼依次用甲苯、水洗涤,干燥后得到29.8 g(98.0%,0.09 mol) 2-(1-氯-环丙-1-基)-1-(苯基)-2-羟基-3-(1,2,4-三唑烷-5-硫羧-1-基)-丙烷,直接用于下一步反应。
2-(1-氯-环丙-1-基)-1-(苯基)-3-(4,5-二氢-1,2,4-三唑-5-硫羰-1-基)-丙-2-醇:在500 mL 四口瓶中加入上一步得到的2-(1-氯-环丙-1-基)-1-(苯基)-2-羟基-3-(1,2,4-三唑烷-5-硫羧-1-基)-丙烷(29.8 g,98.0%,0.09 mol)和甲醇(70.0 g),再于25 ℃左右缓慢滴入氯化铁溶液(127.0 g, 30%,0.20 mol),保温反应5 h。反应结束,过滤,水洗,滤饼用甲醇重结晶得到22.0 g 白色固体(DC-PTC),即2-(1-氯-环丙-1-基)-1-(苯基)-3-(4,5-二氢-1,2,4-三唑-5-硫羧-1-基)-丙-2-醇,含量98.5%,熔点:189~191 ℃。HRMS(m/z,ESI+):310.0774(M+H)+;理论值:310.0775。
1H NMR(400 MHz, DMSO-d6)δ: 8.450 (s, 1H),7.351~7.330 (m, 2H); 7.268~7.200 (m, 3H); 5.091 (s,1H); 4.647 (d,J=14.4 Hz, 1H); 4.328 (d,J=14.0 Hz,1H); 3.171 (d,J=13.6 Hz, 1H); 2.865 (d,J=14.4 Hz,1H); 0.923~0.866 (m, 1H); 0.654~0.596 (m, 1H),0.549~0.400(m,2H)。
1.2.2 2-[2-(1-氯环丙基)-1-(2-氯苯基)-3-(1H-1,2,4-三唑-1-基)丙烷基]-2-醇(DS-PTC)的合成
在500 mL 四口瓶中依次投入1,2,4-三氮唑(27.9 g,99%, 0.40 mol),碳酸钾(55.8 g, 99%, 0.40 mol)和的DMF(150 g),搅拌并升温至90 ℃左右滴加2-(1-氯环丙基)-2-[(2-氯苯基)甲基]环氧乙烷(108.1 g,90%,0.40 mol),滴完后保温反应10 h,然后将反应液降至室温,过滤,滤饼用适量DMF 洗涤,合并DMF溶液,脱溶得到釜残。向其釜残中加入甲苯(200 g)和水(50 g),溶解,分层得到有机相,降温结晶,过滤、干燥得到63.6 g(98.2%,0.20 mol)DS-PTC,熔点:104~106 ℃。HRMS(m/z,ESI+):312.0662(M+H)+;理论值:312.0665。
1H NMR(400 MHz,DMSO-d6)δ: 8.397(s,1H);7.974 (s, 1H); 7.597 (dd,J=7.4, 2.2 Hz, 1H); 7.439(dd,J=7.4,1.8 Hz,1H);7.311~7.211(m,2H);5.145(s,1H); 4.748 (d,J=14.4 Hz, 1H); 4.120 (d,J=14.4 Hz,1H); 3.425 (d,J=14.0 Hz, 1H); 3.116 (d,J=14.0 Hz,1H); 0.898~0.839 (m, 1H), 0.712~0.653 (m, 1H),0.512~0.398(m,2H)。1.2.3 2-{2-(1-氯环丙基)-1-(2-氯苯基)-3[5-(甲巯基)-1H-1,2,4- 三唑-1- 基] 丙烷基}-2- 醇(Me-PCT)的合成
在500 mL 四口瓶中依次投入丙硫菌唑原药(34.8 g,98.8%,0.10 mol)、乙腈(150 g)和无水碳酸钾(13.9 g,99%,0.10 mol),然后于室温、氮气保护下缓慢入碘甲烷(28.7 g,99%,0.20 mol),滴毕体系升温回流反应过夜。反应结束减压蒸馏除去低沸,向其中加入乙酸乙酯(100 g)和水(70 g)充分搅拌溶解,有机相依次用水、饱和食盐水洗涤、无水硫酸钠干燥后旋干得到25.0 g 黏稠油状物,柱层析得到12.5 g 白色固体状甲基化丙硫(98.0%,0.03 mol),熔点60~62 ℃。HRMS (m/z,ESI+):312.0662 (M+H)+;理论值:312.0665。
1H NMR (400 MHz,DMSO-d6)δ:7.999(s,1H),7.676~7.653(dd,J=7.0,2.2 Hz,1H);7.455~7.432(dd,J=7.4,1.8 Hz,1H);7.337~7.265(m,2H);5.078(s,1H);4.560 (d,J=14.0 Hz, 1H); 3.948 (d,J=10.0 Hz, 1H);3.528 (d,J=14.4 Hz, 1H); 3.093(d,J=14.4 Hz, 1H);2.522 (s, 3H); 1.036~0.981 (m, 1H), 0.768~0.647(m,3H)。
2 结果与讨论
2.1 产生DC-PTC 原因分析
原因可能有:⑴起始原料邻氯氯苄中残余少量氯化苄,氯化苄形成的格氏试剂苄基溴化镁依次进行加成、肼化、关环、氧化等后续步骤反应最终生成DC-PTC 杂质;⑵起始原料邻氯氯苄发生格氏反应时,苯环上的氯也参与了格氏反应,加成反应后的酸水中淬灭时,发生水解反应得到脱氯杂质2-苄基-2-(1-氯环丙基)环氧乙烷,脱氯杂质通过后续肼化、合环和氧化等反应最终生成DC-PTC 杂质。反应路线见图3。

图3 DC-PTC 产生原因的路线分析
基于丙硫菌唑登记公开资料,DC-PTC 在其原药中含量约0.5%,而起始原料邻氯氯苄含量>99.5%,据此可以推测,原因1 可能性小。基于原因2,即邻氯氯苄与镁发生格氏反应时,邻氯氯苄苯环上氯也与镁发生格氏反应,由于反应活性较弱,无法与2-氯-1-(1-氯环丙基)乙酮发生反应,在后续酸水中淬灭时形成2-苄基-2-(1-氯环丙基)环氧乙烷,通过后续肼化、合环、氧化等反应最终生成DC-PTC 杂质。
2.2 产生DS-PTC 原因分析
据文献[6]报道,丙硫菌唑采用图4 中R1 路线合成,即化合物1 与1,2,4-三氮唑反应生成DS-PTC,DS-PTC 再与硫磺反应得到丙硫菌唑。该路线收率低,条件苛刻,硫氧化反应不彻底,导致少量的DS-PTC残留。据环境公开数据,DS-PTC 的限量为0.5%。

图4 DS-PTC 产生原因的路线分析
DS-PTC 本身毒性较高,且原研路线残留量较大,很难除去。因此原研公司又开发了路线R2,即化合物1 依次与水合肼、甲醛、硫氰酸钠和氯化铁等反应得到丙硫菌唑。在氯化铁氧化过程中,仍会有极少量的(约100 mg/L)DS-PTC 形成(图4)。
2.3 产生Me-PTC 原因分析
据文献,丙硫菌唑原药合成步骤是在甲醇中进行的,推测存在少量甲醇与丙硫菌唑发生反应形成Me-PTC 的情况。
本文参照现有文献方法对其进行合成,即采用碘甲烷与丙硫菌唑反应制备甲基化丙硫[7],具体的化学反应式如图5 所示。
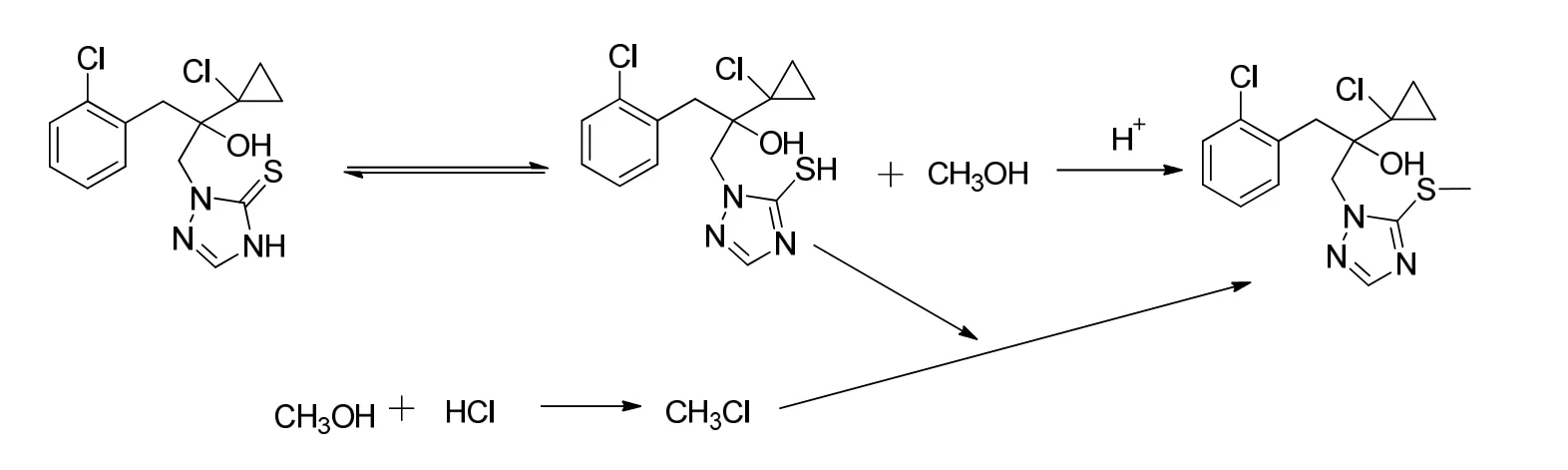
图5 Me-PTC 产生原因的路线分析
3 结论
基于丙硫菌唑原药合成路线和相关文献资料调查,分析推测了丙硫菌唑原药中脱氯丙硫、脱硫丙硫和甲基化丙硫3 个主要杂质产生的可能历程,并设计了合成路线对3 个杂质进行了合成、纯化,合成杂质含量>97%。研究结果可为丙硫菌唑研究、开发提供基础与指导。
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
农药科学与管理(2019年7期)2019-11-29
农药科学与管理(2019年10期)2019-04-20
今日农药(2016年8期)2016-11-15
中国农资(2016年31期)2016-09-13
中国农资(2016年34期)2016-08-11
中国农资(2016年36期)2016-08-09
中国农资(2016年33期)2016-08-02
中国果菜(2016年9期)2016-03-01
西华师范大学学报(自然科学版)(2015年3期)2015-02-27
