
蒽吡唑类衍生物的合成及构效关系
2014-07-02 01:32王建红王朝义张亚宏
河南大学学报(医学版) 2014年3期
王建红,王朝义,陈 冰,高 凡,张亚宏,赵 瑾
(河南大学河南省天然药物与免疫工程重点实验室,河南开封475004)
蒽吡唑类衍生物的合成及构效关系
王建红,王朝义,陈 冰,高 凡,张亚宏,赵 瑾*
(河南大学河南省天然药物与免疫工程重点实验室,河南开封475004)
目的 以蒽醌为原料,与脂肪胺通过缩合反应合成系列蒽醌-胺基衍生物。方法 利用1H NMR、MS、元素分析等方法对所合成的目标化合物进行了结构确认。结果 肿瘤细胞生长抑制实验结果表明,该类蒽吡唑衍生物在相应的细胞模型显示出一定的肿瘤细胞生长抑制能力。利用髙亚精胺修饰的蒽醌衍生物对所测试细胞具有显著的生长抑制活性。结论 初步的构效关系分析发现,蒽吡唑母核C-3位的化学修饰对提高蒽咇唑衍生物的抗肿瘤活性影响不大,然而侧链取代基对于改善其抗肿瘤作用具有显著影响。特别是利用多胺链修饰得到的衍生物不但能够有效提高抗肿瘤作用,还对耐药细胞SMMC-7721(耐药人肝癌细胞)表现较强的生长抑制作用。
蒽吡唑;结构修饰;合成;抗肿瘤活性
蒽咇唑类化合物在药物研究中具有重要地位。其杂合了蒽醌骨架和咇唑环,蒽醌结构具有DNA嵌入作用,是临床药物中常见的母核。以蒽醌为基础的药物,在临床上用于急性白血病和各种实体瘤[1-4]。但这些药物普遍具有心脏毒性而限制了它们的广泛应用[2-5]。
近年来,药物研究工作者[6-7]发现基于蒽醌母核的蒽咇唑结构具有较低的心脏毒性和良好的抗肿瘤作用。围绕蒽咇唑开发的药物,如洛萨蒽醌(Losaxantrone)和吡罗蒽醌(Piroxantrone)已经应用于临床,并具有良好的治疗效果。
以蒽咇唑为基础,围绕其母核和侧链进行结构修饰获得目标化合物,通过对其进行体外抗肿瘤活性评价,研究该类型的结构改造对蒽咇唑类衍生物生物活性的影响,以期发现良好的药物先导化合物。目标化合物通过图式1和2的方法合成。

图1 化合物4a,4b的合成路线
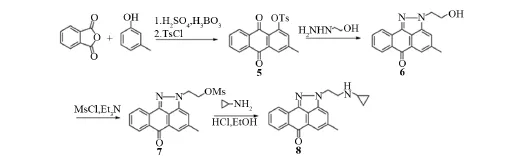
图2 化合物8的合成路线
1 实验部分
1.1 仪器与试剂
X-6精密熔点测定仪(温度未经校正)、VarianINOVA-400型核磁共振仪、Esquire 3000型LC-MS质谱仪、Vario ELⅢ型元素分析仪。
化学合成中的试剂均为市售分析纯,使用前未经提纯处理。
1.2 目标化合物4a、4b和8的合成通法
将化合物3(0.36 g,1.0 mmol)溶于20 m L无水乙醇中,加入相应的环丙基胺、髙亚精胺 (20 eqiv.),加热回流约8 h,TLC控制原料基本反应完全,减压蒸除溶剂,残余物用柱层析分离提纯(洗脱剂比例为氯仿/甲醇=100/1~100/5),得到中间体。将中间体溶于适量乙醇中,并在冰水浴条件下逐滴加入盐酸乙醇溶液(4 mol/L),加毕自然升至室温,搅拌反应过夜,有沉淀生成,离心,除去上层液体,依次用乙醇、乙醚清洗直至上层液体为亮黄色。收集固体干燥后得最终产物4a,4b。
4a:浅黄色固体,产率60.1%,1H NMR(D2O, 400 MHz)δ:7.51(d,1H,J=4.0 Hz,Ar H), 7.43(t,2 H,J=5.0 Hz,Ar H),7.317.22(m, 3H,Ar H),7.16(d,1H,J=4.0 Hz,Ar H),4.53 (t,2H,J=6.0 Hz,CH2),3.71(t,2H,J=6.0 Hz,CH2),2.882.83(m,1 H,,CH),1.030.92 (m,4 H,2CH2).C19H18Cl N3O·0.4H2O元素分析实测值(计算值)%:C 65.81(65.76),H 5.31 (5.46),N 12.07(12.11).MS(ESI)m/z:304.3(M +H)+.
4b:黄色固体,产率48.2%,1H NMR(D2O,400 MHz)δ:7.38~7.30(m,3H,Ar H),7.20~7.19( m,2 H,Ar H),7.12(s,1 H,Ar H),7.03(s,1 H, Ar H),4.45(t,2 H,J=6.0 Hz,CH2),3.57(t, 2H,J=6.0 Hz,CH2),3.30(t,2 H,J=8.0 Hz, CH2),3.21(q,4 H,J=8.0 Hz,2CH2),3.12(t, 2H,J=8.0 Hz,CH2),2.212.03(m,4H,2CH2). C22H30Cl3N5O·3.5H2O元素分析实测值(计算值)%:C 48.07(48.05),H 6.77(6.78),N 12.78 (12.74).MS(ESI)m/z:378.5(M+H)+.
8:淡黄色固体,产率62.1%,1H NMR(D2O, 400 MHz)δ:7.75(d,J=8.0 Hz,1 H,Ar H), 7.61(d,J=8.0 Hz,1H,Ar H),7.537.48(m, 2H,Ar H),7.19(s,1H,Ar H),7.01(s,1H, Ar H),4.51(t,2 H,J=6.0 Hz,CH2),3.70(t,2H,J=6.0 Hz,CH2),2.90(m,1H,CH),2.33 (s,3 H,CH3),1.00(m,4 H,2CH2).C20H24Cl N3O·1.2H2O元素分析实测值(计算值)%:C 64.13(63.98),H 5.98(6.01),N 10.99(11.19). MS(ESI)m/z:318.4(M+H)+.
1.3 肿瘤细胞生长抑制活性测试
取对数生长周期的肿瘤细胞,调整细胞数为5× 103个/m L加于96孔培养枚中,每孔100μL。分样品组、对照组(不加样品)和空白组,每组设定4个复孔。于饱和湿度、体积分数5%CO2、37℃条件下培养48 h,每孔加入5 mg/m L的MTT 10μL,继续培养4 d后每孔加入10%十二烷基硫酸钠溶液(悬浮细胞)或 DMFO(贴壁细胞)100μL,过夜,于570 nm处测定吸光度值。由公式:抑制率=(A对照-A样品)/(A对照-A空白)×100%来计算不同浓度的抑制率。
2 结果与讨论
2.1 活性测试结果
采用MTT法测试了目标化合物对Hep G2(人肝癌细胞);BEL-7402(人肝癌细胞);HCT-116(人结肠癌细胞);HT-29(人结肠癌细胞);SMMC-7721 (耐药人肝癌细胞)五种细胞的生长抑制活性,并用临床药物米托蒽醌作参照药物。在37℃下培养48 h,目标化合物的细胞生长抑制浓度(IC50)结果见表1。

表1 目标化合物对肿瘤细胞的生长抑制活性
2.2 构效关系分析
从目标化合物的细胞生长抑制活性结果(表1)可以看出,化合物4a和8对所测试细胞的抑制活性均大于50μM,低于参照化合物米托蒽醌。而化合物4b则显示出显著的细胞毒性,与米托蒽醌相比,4b在相应的细胞模型上IC50远远小于米托蒽醌,显示出较强的肿瘤细胞生长抑制能力。特别是4b对耐药细胞SMMC-7721(耐药人肝癌细胞)也表现较强的生长抑制作用(IC50=10.3μM),提示出该类结构在克服耐药性方面的潜在应用。
结合目标化合物的结构来看,我们合成的化合物结构均以蒽咇唑母核为基础进行结构修饰,然而不同的结构改变显示出截然不同的生物活性。例如化合物4a和8,结构上只有蒽咇唑母核C-3位取代基不同,但是没有导致肿瘤细胞生长抑制活性的差别,该结果提示C-3位的结构修饰对其生物活性影响较小。而化合物4a和4b相比,它们的母核相同,结构上只有侧链取代基不同(4a为环丙基取代,4b为高亚精胺基),但是二者的活性测试结果显示出明显的差别。与4a相比,4b对所测试的细胞模型均有较强的生长抑制作用,重要的是4b对耐药细胞也显示较强的抑制能力。该结果表明,蒽咇唑侧链的胺基取代一方面有利于提高抗肿瘤活性,另一方面也有助于化合物克服肿瘤细胞的耐药性。
[1]Vuimo T A,Kulikova E V,Sinauridze E I,et al.New research on biotechnology and medicine[M].New York: Nova Science Publishers,2006:300.
[2]Monneret C.Recent developments in the field of antitumor anthracyclines[J].Eur J Med Chem,2001,36(6):483-493.
[3]Faulds D,Balfour J A,Chrisp P,et al.Mitoxantrone,a review of its pharmacodynamic and pharmacokinetic properties,and therapeutic potential in the treatment of cancer [J].Drugs,1991,41(3):400-449.
[4]Wiseman L R,Spencer C M.Mitoxantrone.A review of its pharmacology and clinical efficacy in the management of hormoneresistant advanced prostate cancer[J].Drugs Aging,1997,10(6):473-485.
[5]Tarasiuk J,Mazerski J,Tkaczyk-Gobis K,et al.Molecular basis of the low activity of antitumor anthracenediones,mitoxantrone and ametantrone,in oxygen radical generation catalyzed by NADH dehydrogenase.Enzymatic and molecular odeling studies[J].Eur J Med Chem, 2005,40(41):321-328.
[6]Judson I R.Anthrapyrazoles:true successors to the anthracyclines?[J].Anticancer Drugs,1991,2(3):223-231.
[7]Fry D W.Biochemical pharmacology of anthracenediones and anthrapyrazoles[J].Pharmacol.Ther,1991,52(1):109-125.
[责任编辑 李武营]
Synthesis and structure activity relationship of anthrapyrazoles derivatives
WANG Jianhong,WANG Zhaoyi,CHEN Bing,GAO Fan,ZHANG Yahong,ZHAO Jin*
(Key Laboratory of Natural Medicines and Immunotechnology of Henan Province,Henan University,Kaifeng,Henan 475004,China)
Objective Several anthrapyrazoles derivatives were synthesized using anthraquinone and aliphatic amines as starting material.Methods The structure of the target compounds were confirmed by 1H NMR,MS and elemental analysis assays.Results The biological results indicated that these derivatives exhibited potent in vitro cytotoxicity against different cancer cell lines.Among them,the polyamine based anthrapyrazole derivatives showed superior cytotoxicity than that of Mitoxantrone both on cancer cell lines and the drug resistant subline.Conclusion The structure activity relationship of these anthrapyrazoles derivatives demonstrated that the polyamine side chain could be helpful in improving the antitumor activity,while the C3 site on anthrapyzole had little effect on the cytotoxicity.
Anthrapyrazole;Structural modification;Synthesis;Antitumor activity
O 625.46
A
1672-7606(2014)03-0164-03
2014-03-26
国家自然科学基金(21272056);河南省科技厅基础与前沿项目(112300413224);河南省教育厅自然科学项目(2010B350002)。
王建红(1971—),男,河南尉氏人,副教授,从事化学与药物合成的研究工作。
*通讯作者:赵瑾(1948-),女,河南开封人,教授,博士生导师,从事天然产物与药物研究工作。
猜你喜欢
今日农业(2021年2期)2021-11-27
今日农业(2020年23期)2020-12-31
世界农药(2019年3期)2019-09-10
中成药(2018年10期)2018-10-26
天然产物研究与开发(2018年8期)2018-09-10
中成药(2018年4期)2018-04-26
中学生数理化·高二版(2016年3期)2016-12-26
天然产物研究与开发(2016年11期)2016-06-15
合成化学(2015年1期)2016-01-17
合成化学(2015年10期)2016-01-17
