
基于Cytoscape的蛋白质网络可视化聚类分析插件
2014-11-14 07:10唐羽,李敏
生物信息学 2014年1期
唐 羽,李 敏
(中南大学信息科学与工程学院,长沙410083)
蛋白质是生物完成各种生命活动,实现各种生命功能所必需的大分子物质。生物体的各种功能并不是通过单个蛋白质表现出来,而是通过众多蛋白质之间在特定条件下的相互作用才能表现出一定的功能。生物系统是由许多相互作用的、相对独立的结构化功能模块组成,识别出这些模块对于理解生物系统的组织结构具有重要意义。聚类分析是识别这些功能模块的有效手段。
蛋白质网络可视化对于更快速,更有效,更直观的分析蛋白质网络特性起到了重要的作用。尤其是对蛋白质网络作聚类分析的时候,聚类分析结果的可视化处理无疑将有利于更快速地得出正确结论。因此,本文将蛋白质网络的聚类分析和生物网络可视化功能相结合,开发了一个集成于Cytoscape[1]的蛋白质网络聚类分析和显示插件CytoCluster。
本插件不仅集成了 MCODE[2],FAG - EC[3],HC-PIN[4],OH -PIN[5],IPCA[6],EAGLE[7]等六种典型的聚类算法,实现了使用多种算法对网络进行聚类分析,而且还提供了聚类结果可视化功能,能,将分析所得的clusters以缩略图列表的形式直观地显示出来,对于单个cluster,可显示在原网络中的位置,并能生成相应的子图单独显示,有助于研究人员对cluster功能特性更深入研究。CytoCluster创造了一个更快速,更有效,更直观的分析蛋白质网络特性的研究环境,可为生物学家提供更加有价值的参考信息。
1 系统原理及总体结构
1.1 系统功能模型
本系统旨在基于Cytoscape这个可视化平台对蛋白质网络进行聚类分析,系统功能模型分为了聚类分析与界面控制两大模块,其中界面控制部分由Bundle控制、面板控制、聚类结果可视化、结果排序、导出结果等五个子模块构成。如图1所示。

图1 系统功能模型图Fig.1 System function model
1.2 系统集成的聚类算法
本系统集成了 MCODE、FAG-EC、HC-PIN、OH-PIN、IPCA、EAGLE等六种典型的网络聚类算法,具体介绍如下。
MCODE(The molecular complex detection algorithm)算法是一种基于密度的非交叠式聚类算法。该算法以种子节点为中心进行扩展在其邻居节点中寻找满足要求的节点,从而形成一个功能模块,最早在Bader etal的早期文章以及文献[8]中被提出,针对通过构建cluster来在蛋白质相互作用网络中检测复合物。其核心思想是以局部密度所定义的adhoc网络为根据,分离局部稠密区域。
FAG-EC(Fast agglomerate algorithm)算法是基于边聚类系数(Edge clustering coefficients)的非交叠式聚类算法,速度上的优越性比较明显。该算法采用自底向上的凝聚算法进行模块识别,并提出了一个新的参数化的模块定义。通过将按聚集系数非增序排列的边序列逐条加入初始化为单个节点的各个模块中来合并各个团,直到达到定义的模块要求。
HC-PIN(Fast hierarchical clustering algorithm)算法是一个快速层次非交叠式聚类算法,由FAGEC算法改良而来,同样采用自底向上的凝聚算法进行模块识别,不同的是,该算法以边聚类值(Edge clustering value)为基础,即可应用于无权网络也可用于加权网络的聚类分析。
OH-PIN(Identification of hierarchical and overlapping functional modules)算法是可识别层交叠蛋白质功能模块的凝聚式层次算法,以M_clusters,λ-module,以及cluster间的聚集系数为基础进行聚类计算,在功能富集化以及比较已知蛋白质复合物方面,性能优于较为优越。
IPCA(Clusteralgorithm based on the new topological structure)算法由 DPClus[9]算法改良而来的基于密度的算法。该算法利用一种新型拓扑结构来预测网络中蛋白质复合物,以子图直径(或节点平均距离)和子图密度来对识别过程进行控制调节,可识别交叠的蛋白质功能模块,在寻找已知蛋白质复合物方面,性能表现良好。
EAGLE(Agglomerative hierarchicalclustering based on maximal clique)算法,是一种可以识别交叠功能模块的凝聚式层次算法,以极大团为基础,通过逐步合并相似性最大的两个团,找到最优划分方法来实现的。
1.3 系统主要功能模块
(1)聚类分析模块:实现 MCODE,FAG-EC,HC-PIN,OH-PIN,IPCA,EAGLE等六种算法的具体分析过程。初步得出聚类分析结果。
(2)Bundle控制模块:由于Cytoscape3.0采用了OSGi进行架构,因此在Cytoscape平台上运行的大型插件只能以Bundle Apps的形式出现。该模块实现CytoCluster激活关闭服务调用等功能。
(3)面板控制模块:该模块负责插件整体界面实现,由APP菜单、参数面板、各个算法面板以及聚类结果面板等子模块构成。
(4)聚类结果可视化模块:该部分将初步聚类分析结果可视化的呈现出来,主要包括聚类结果列表中cluster缩略图的实现,显示单个cluster在原网络中的位置,以及生成并单独显示被选中的cluster子图。
(5)导出结果模块:负责对聚类结果进行导出,对本次分析使用的算法、参数以及所得的cluster的具体信息进行了详细地记录。
2 系统设计与实现
2.1 系统主要数据结构
为了实现对待分析网络聚类分析识别出功能模块,并将识别结果显示给用户,并提供进一步的分析处理,CytoCluster系统类图如图2。需要用到的类和对象如下:
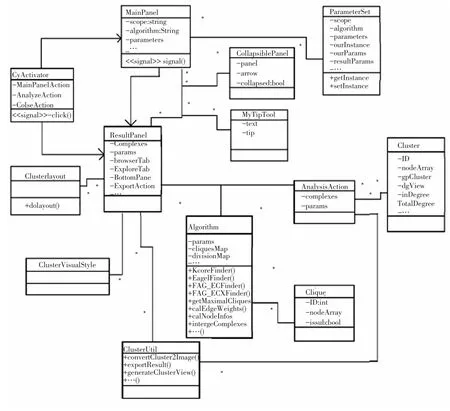
图2 CytoCluster类图Fig.2 Class diagram of CytoCluster
(1)CyActivator:系统入口,将插件在OSGi框架中激活或关闭,调用系统服务等。
(2)MainPanel:程序主面板。
(3)ResultPanel:结果显示面板。
(4)Algorithm:程序聚类算法实现类。
(5)AnalyzeAction:实现对analyze按钮的响应,调用AnalyzeTaskFactory开始聚类分析
(6)AnalyzeTaskFactory:该类用于产生AnalyzeTask。
(7)AnalyzeTask:该类是聚类分析的入口,负责实现用户选择的聚类算法
(8)MyTipTool:可显示多行文字的提示工具。
(9)CollapsiblePanel:可折叠的面板。
(10)Cluster:CytoCluster使用的表示一个功能模块所采用的数据结构。
(11)Clique:CytoCluster中表示极大团所采用的数据结构。
(12)ParameterSet:聚类分析时所用的参数集合。
(13)ClusterLayout:CytoCluster使用的节点布局,在ClusterUtil中生成模块图像和创建子图时用到。
(14)ClusterVisualStyle:CytoCluster使用的视图显示风格,在ClusterUtil中生成模块图像
(15)ClusterUtil:包含对得到的功能模块集合Cluster[]进行的各种处理函数集合。
2.2 界面控制模块实现
(1)Bundle控制
该模块主要通过实现了 org.cytoscape.service.util.AbstractCyActivator接口的 Cyactivator类完成,实现了OSGi框架中服务注册,Bundle激活以及关闭等功能。
若要调用OSGi框架中现有的服务,需要调用Cyactivator类中的 getService(Bundle Context bc,Class<CyApplicationManager> serviceClass)方法。
若要将自己写的类注册为框架中的服务,需要调用 Cyactivator类中registerService(Bundle Context bc, Object service, Class <? > serviceClass,Properties props)方法,如图3所示。
Bundle激活以及关闭部分主要通过OpenTaskFactory类以及CloseTaskFactory类完成。
(2)面板控制
插件整体界面由APP菜单、各参数入面板以及聚类结果面板等子模块构成。插件最终运行整体效果图如图3所示。

图3 CytoCluster运行效果Fig.3 The running interface of CytoCluster
参数输入面板用于输入聚类分析所需各项参数。启动聚类插件后显示主面板后,用户按需要选取分析范围,选择聚类算法:
MCODE、FAG -EC、HC -PIN、OH -PIN、IPCA、EAGLE中的一种。用户选择算法后,在下方显示相应的参数选项,用户可直接使用默认参数或自定义各参数值。参数设定完成后单击Analyze按钮,首先进行参数有效性判断,参数无误后则按照用户定义的参数进行开始分析过程。
聚类结果面板用于显示识别出的所有功能模块信息,并提供其它一些控件来对聚类分析结果作进一步的分析处理。
如图3所示,A、B、C分别为参数输入面板,可视化面板,聚类结果面板;a部分为分析区域选择,b部分为算法种类选择,c部分为算法参数面板;d部分为BrowserTable,e部分为选中的 cluster,j部分为模块尺寸滑动条,f部分为cluster节点属性显示列表,g为创建 cluster子图按钮,h关闭结果面板按钮,i为导出聚类结果按钮。
BrowserTable表格分为两列,分别显示了功能模块缩略图形以及此功能的基本信息:模块中的节点数,边数以及分值等信息。CytoCluster还提供对聚类结果进行排序的功能。排序时有三种不同的排序方式可供选择:按模块大小,按模块的模块性以及按照模块的score值进行排序。其中,按score排序只在使用MCODE算法进行聚类时有效,其它算法中模块的score值全部为0。
节点属性列表显示了由Cytoscape载入网络时读入的当前网络中所有包含的属性值,用户可以从下拉列框中选择具体某项,属性列表中就相应地显示当前模块中具有该属性的所有属性值出现的次数分布情况。Create SubNetwork按钮可以将当前模块建立为一个新的网络,并显示于桌面。
另外,当使用MCODE算法进行聚类时,当前模块属性面板中增加了一个模块大小控制面板,其中有一个滑动条,用滑块位置控制增大或缩小当前功能模块的尺寸大小,并对应更新显示模块缩略图。
ResultPanel底部面板由两个按钮组成:Export按钮用于将当前聚类结果中识别的功能模块导出为文件,可以选择导出基本信息和导出完整信息两种方式。Dicard按钮用于在向用户确认之后关闭当前打开的结果面板。
(3)聚类结果可视化
该模块分为三个主要任务,聚类结果列表中cluster缩略图的实现,显示单个cluster在原网络中的位置,以及生成并单独显示被选中的 cluster子图。
cluster缩略图主要通过ClusterUtil类中convertClusterToImage方法来实现。该方法首先通过 ClusterUtil类中 createSubNetwork(CyNetwork net,Collection nodes,SavePolicy policy)方法来为分析所得的 cluster创建子图,再根据这个子图调用createNetworkView(CyNetwork net,Visual-Style vs)方法创建相应的视图,其中VisualStyle为视图属性对象,通getClusterStyle()获得。在这个方法中,设置了视图中节点的大小、颜色,边的颜色、粗细等属性,基本确定了缩略图的显示风格。
得到视图对象后,调用Cytoscape所提供的可视化 接 口 org.cytoscape.view.presentation 的createImage(Int width,Int height)方法生成image对象返回给结果面板声称对象。
cluster子图生成并显示的过程与生成cluster缩略图较为相似,首先调用createSubNetwork创建子图对象,再通过 getClusterStyle设置视图显示风格,createNetworkView生成相对应的子图视图。但与生成缩略图不同,该任务主要是要将生成的子图显示在Cytoscape网络放大显示在面板上,这主要通过ClusterUtil类中的displayNetworkView(CyNetworkView)方法实现。
(4)导出结果
该模块由结果面板中的Export按钮触发调用,将最终聚类结果,即各个cluster的名称、节点总数、节点名称等信息以文本形式导出存入text文件。
该模块主要通过ClusterUtil类中的exportResults方法实现。
其中,FileUtil是 Cytoscape系统提供的文件打开接口,通过调用该接口中的getFile方法,返回新创建的输出文件。FileChooserFilter为文件类型选择器,通过该类存储文件类型,设为txt文本文档。通过FileWriter类将需要输出的cluster数据,如各个cluster的名称、节点总数、节点名称等信息写入文件中。
2.3 聚类分析模块实现
CytoCluster中的聚类算法实现部分,所有的聚类算法都是通过调用一个Algorithm对象的相应方法来完成。
MCODE算法使用K_CoreFinder()识别功能模块。Algorithm对象调用此方法前须先调用scoreGraph方法对网络图中的各个节点的计算MCODE算法所需的节点信息:包括自身在内的邻居接点子图neighbors及其密度density,以该点为种子节点所能扩展出的最大k值的K-Core,其k值水平coreLevel,此K-Core的密度coreDensity以及该节点的score值。节点的score值反映了该节点及其周边节点的密集程度。然后再从score值最大的节点开始,调用getClusterCore()方法,以此节点为种子节点开始扩展,逐步加入符合参数条件的邻接节点。最后根据参数要求作一些后续处理,得出最终的功能模块。
FAG_EC算法使用FAG_ECFinder()识别功能模块。计算的先决条件必须先调用calEdgeWeight得到网络中所有边的聚集系数,并按非增序排列。这里得到的边聚集系数队列也可以多次重复使用。然后先将网络中每个节点初始化为一个Complex,而后开始逐步将各条边依次加入各Complex,从而对Complex进行合并,逐渐成为不可合并的功能模块,直到所有边都被加入 Complex为止。FAG_ECXFinder()使用扩展的FAG_EC算法识别功能模块。首先同EAGLE一致,调用 getMaximalCliques,以极大团为基础得到初始的Complexes集,同时需得到网络中的按聚类系数排列的边的非增序列,调用calEdgeWeight。根据边两端点所从属的Complex集的关系,对两组Complex分别合并后再合并为一个Complex,中途如果有的Complex已达到功能模块定义,则不将之合并。反复进行这个过程,直到所有边都被已处理完为止。
HC-PIN算法使用HCPIN Finder()识别功能模块。计算的先决条件必须先调用calEdgeWeight得到网络中所有边的聚集值,并按非增序排列。这里得到的边聚集系数队列也可以多次重复使用。然后先将网络中每个节点初始化为一个Complex,而后开始逐步将各条边依次加入各Complex,从而对Complex进行合并,逐渐成为不可合并的功能模块,直到所有边都被加入Complex为止。
OH-PIN算法使用OHPINFinder()识别功能模块,首先调用calB_cluster按网络中的边得到相应的B_cluster,再用calC_set将 B_cluster存入 C_set中。通过calOS计算出C_set中cluster间的Overlapping Score,取值最大的一对cluster,合并,直到所有值小于给定的 os_th;再通过 calCCV计算出 C_set中cluster间的CCV,取值最大的一对cluster,合并,直到所有值小于0为止;
IPCA算法使用IPCAFinder()识别功能模块。计算的先决条件必须先调用calNodeWeight得到网络中所有节点的权值,并按非增序排列。选出最大的节点为种子,通过ExtendingCluster来进行扩展,其中用SPJudgement来判断某个点是否该加入当前的cluster。当所有可能被加入的节点都被探测过以后,输出 cluster.
EAGLE算法使用EAGLEFinder()识别功能模块。EAGLE算法基于极大团,故Algorithm对象必须先调用getMaximalCliques方法计算出网络中的所有极大团。然而一个网络中的极大团是固定不变的,所以计算一次后就可以供以后需要时使用。接下来首先完成团初始化工作,去除附属极大团。开始合并后,每次选出相似性最大的两个Complex将之合并,并调用calModularity计算当前划分优劣程度的EQ值。重复合并过程直到只有一个Complex,
并记录下整个过程中每一次的划分情况及其EQ值。选出其中EQ的最大值,此时的团划分情况即是最优的功能模块划分,返回这个模块集合。
3 实例展示及分析
3.1 聚类结果可视化
CytoCluster不仅能对网络进行聚类分析,同时也实现了对网络聚类结果的可视化显示,将分析所得的clusters以缩略图列表的形式直观地显示出来,对于单个cluster可显示在原网络中的位置,并能生成相应的子图单独显示,如图4所示。通过该功能,研究人员可以对基因以及蛋白质网络中分子簇的进行进一步挖掘,发现致病基因以及蛋白质功能模块的相关特性以及相互关联性。将聚类分析所得的cluster放在统一网络中相互比较,展示相互之间的关联性,并可由用户自行设定节点的颜色和形状,对于研究网络中的功能模块具有重要作用。例如宾夕法尼亚大学医学院的研究人员利用FAG-EC算法对注意力缺陷多动障碍症进行研究时,根据网络的拓扑结构,将整个基因网络聚类成17个不同的分子簇[10],显示簇内节点以及簇与簇之间的关系。

图4 聚类结果可视化实现效果Fig.4 The implementation of visualization clustering result
3.2 聚类结果对比与分析
图5为Cytocluster中六种聚类算法对Cytoscape所提供的酵母核心蛋白质相互作用网络(GalFiltered.sif)的分析结果,由图可知CytoCluster所采用的六种聚类算法得到的结果之间存在一定的差异。在具体使用中可以结合多种算法进行聚类分析,从而得到对蛋白质网络中的功能模块的更全面认识。
为了分析比较CytoCluster中实现的几种聚类算法的性能,这里使用从数据库DIP获得的酵母核心蛋白质相互作用网络数据集Y2k进行聚类分析,得出6种不同的功能模块数据,对这些模块集进行过滤,除去尺寸小于3的模块,再查找这些功能模块的GO Terms。
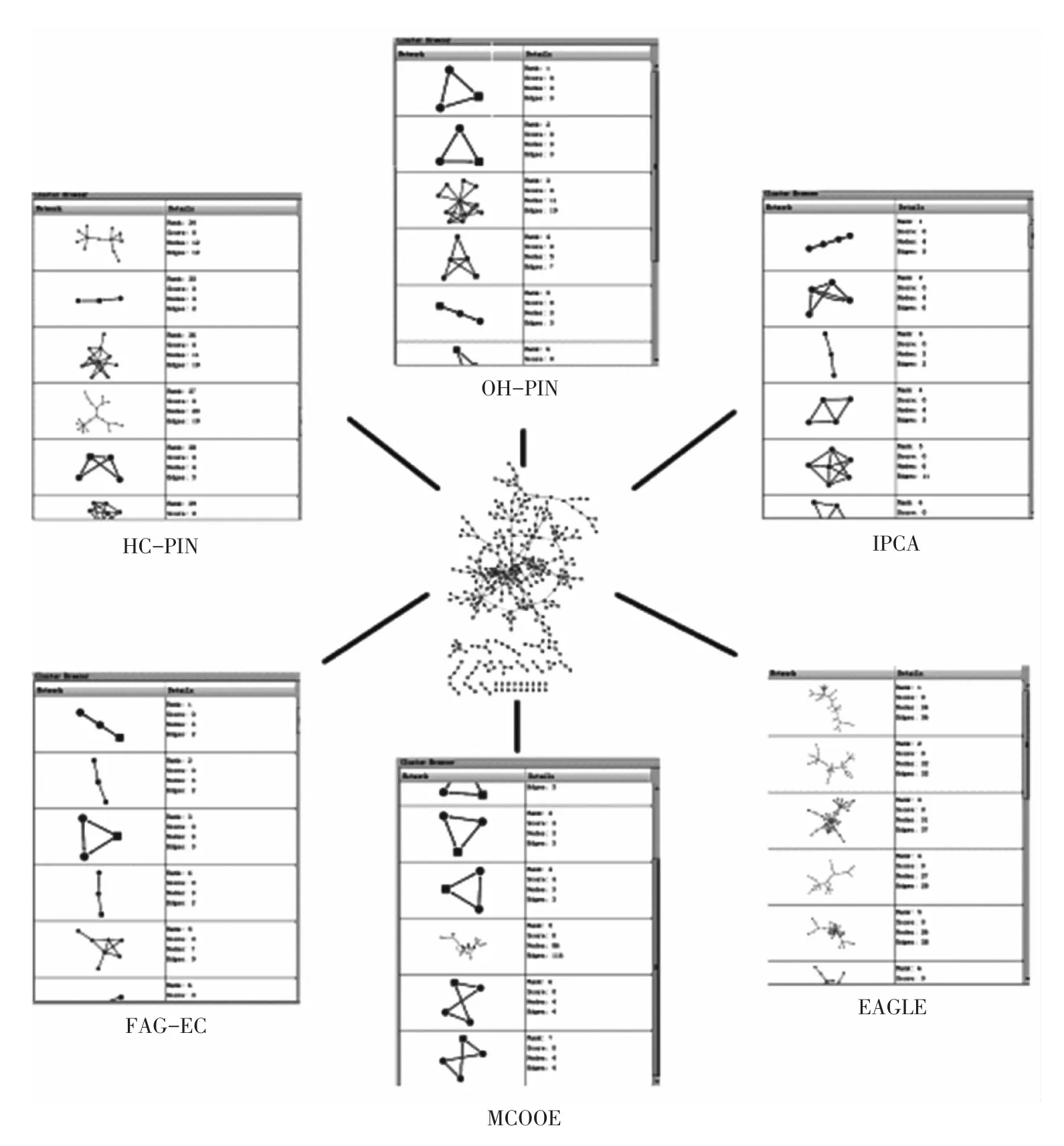
图5 六种算法对同一网络聚类分析对比Fig.5 The comparison results of different clustering algorithms
由于所得的数据繁多,这里仅采用GO中的生物过程Process为例,对各功能模块集合的富集性P-value值分布进行分析,结果如图6所示。
由于在统计检验中,P-value值可用于评价一组蛋白质集合在偶然情况下聚集成一个模块的可能性大小,所以从图中可以看出,各聚类算法在识别的功能模块结构和数量上存在不小的差异,但这些模块在性能、准确度上各有所长,对更全面地认识整个蛋白质网络具有重要意义。

图6 不同聚类算法识别模块的对应富集性P-value分布Fig.6 The enrichment analysis result of different algorithm
4 讨论
CytoCluster实现了在Cytoscape3.0平台上使用了 MCODE,FAG -EC,HC -PIN,OH -PIN,IPCA,EAGLE等六种典型聚类算对蛋白质网络进行聚类分析,并进一步对聚类结果可视化显示分析与比较。
本插件的实现总体来说可概括为以下两部分:
(1)后台蛋白质聚类算法的实现。这是本插件的核心部分,由于旨在于Cytoscape这个可视化平台上对蛋白质网络进行聚类分析比较从而为生物信息学研究人员提供一个直观准确的具有参考价值的结果,因需要十分注意软件的精准性、可靠性与严谨性,每个聚类算法要有据可查,要严格的与原算法过程及结果保持一致。
(2)前台界面控制与可视化的实现。包括Bundle控制,面板控制,聚类结果可视化,导出结果等部分,是该插件有别于其他简单聚类分析软件的重要区别所在,使得该软件不仅能对蛋白质网络进行聚类分析,还能进一步可视化聚类结果进行分析比较,并导出到文本中。
CytoCluster系统是一个可扩展平台,随着蛋白质网络聚类技术的发展,将在今后的开发中集成更多的聚类算法,使得系统更加丰富,更加完善。
References)
[1] SHANNON P,MARKIEL A,OZIER O,et al.Cytoscape:a software environment for integrated models of biomolecular interaction networks[J].Genome Research,2003,13(11):2498-504.
[2] BADER G D,HOGUE C W V.An automated method for finding molecular complexes in large protein interaction networks[J].BMC bioinformatics,2003,4(1):2.
[3] LI Min,WANG Jianxin,CHEN Jianer.A fast agglomerate algorithm for mining functional modules in protein interaction networks[C]//BioMedical Engineering and Informatics,2008.BMEI 2008.International Conference on.IEEE,2008,1:3-7.
[4] WANG Jianxin,LI Min,CHEN Jianer,et al.A fast hierarchical clustering algorithm for functional modules discovery in protein interaction networks [J].Computational Biology and Bioinformatics,IEEE/ACM Transactions on,2011,8(3):607-620.
[5] WANG Jianxin,REN Jun,LI Min,et al.Identification of Hierarchical and Overlapping Functional Modules in PPI Networks[J].IEEE TRANSACTIONS ON NANOBIOSCIENCE,2012,11(4):386 -393.
[6] LI Min,CHEN Jun,WANG Jianxin,et al.Modifying the DPClus algorithm for identifying protein complexes based on new topological structures [J]. Bmc Bioinformatics,2008,9(1):398.
[7] SHEN Huawei,CHENG Xueqi,CAI Kai,et al.Detect overlapping and hierarchicalcommunity structure in networks[J].Physica A:Statistical Mechanics and its Applications,2009,388(8):1706-1712.
[8] RADICCHI F,CASTELLANO C,CECCONI F.Defining and identifying communities in networks [J].Proc.Natl.Acad.Sci.USA,2004,101(9):2658 -2663.
[9] ALTAF-Ul-AMIN M,SHINBO Y,MIHARA K,et al.Development and implementation of an algorithm for detection of protein complexes in large interaction networks[J].BMC bioinformatics,2006,7(1):207.
[10] ELIA J,GLESSNER J T,WANG K,et al.Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder[J].Nature genetics,2011,44(1):78-84.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
机械工业标准化与质量(2021年10期)2021-11-19
海洋信息技术与应用(2020年1期)2020-06-11
传媒评论(2019年4期)2019-07-13
模具制造(2019年4期)2019-06-24
摄影之友(影像视觉)(2017年1期)2017-07-18
软件导刊(2016年9期)2016-11-07
通信电源技术(2016年5期)2016-03-22
石油知识(2016年2期)2016-02-28
自动化仪表(2015年11期)2015-04-01
