
梁山慈竹高通量转录组测序及差异表达基因分析
2016-07-28 01:40王身昌胡尚连
华北农学报 2016年3期
王身昌,胡尚连,曹 颖,徐 刚
(1.西南科技大学植物细胞工程实验室,四川绵阳 621010;
2.四川省生物质资源利用与改性工程技术研究中心,四川绵阳 621010)
梁山慈竹高通量转录组测序及差异表达基因分析
王身昌1,2,胡尚连1,2,曹 颖1,2,徐 刚1,2
(1.西南科技大学植物细胞工程实验室,四川绵阳 621010;
2.四川省生物质资源利用与改性工程技术研究中心,四川绵阳 621010)
分析梁山慈竹及其体细胞突变体的转录组,挖掘功能基因并对其差异表达基因进行筛选和分析,为梁山慈竹遗传改良提供理论依据。利用RNA-Seq技术进行转录组测序,对测序结果进行de novo拼接和功能注释;并对差异表达基因进行筛选及COG、GO、KEGG数据库中进行比对注释,此外,基于Swiss-Prot功能注释结果,分析纤维素和木质素相关功能基因的表达量差异。测序结果表明,共获得86 575 631条reads,de novo组装得到84 741条unigenes,共有49 829条被Nr、COG、GO、KEGG、Swiss-Prot注释。从梁山慈竹实生植株(对照)和体细胞突变体No.30这2个测序样本中,筛选出3 572条差异表达unigenes,757条差异表达unigenes在COG分类体系中具有详细的蛋白功能释义,2 213条差异表达unigenes在GO数据库具有功能定义,385条unigenes被注释到94条KEGG Pathways中。纤维素合成相关纤维素合酶、过氧化物酶、泛素连接酶和热休克蛋白在梁山慈竹体细胞突变体No.30中表达量升高,木质素合成相关MYB4、4-香豆酸CoA连接酶、肉桂醇脱氢酶、肉桂酰-CoA还原酶和漆酶在突变体中表达量降低。提供了全面的梁山慈竹转录组信息,获得了一批在梁山慈竹纤维素和木质素生物合成过程中有重要功能的基因序列。
梁山慈竹;体细胞突变体;转录组;差异表达基因
转录组(Transcriptome)能够从整体水平研究基因功能以及基因结构,揭示特定生物学过程中的分子机理[1]。转录组高通量测序技术已经成功应用于多个非模式生物的研究,如蓝莓经高通量测序筛选得到大量与抗氧化剂合成相关的候选基因,并基于果皮和果肉2个测序数据库中差异表达基因,初步分析与类黄酮抗氧化剂合成相关基因以及转录因子[2];麻竹经转录组测序技术,筛选出与木质素合成、生长发育相关基因以及转录因子,为麻竹基因组学研究提供非常有价值的资源[3]。
梁山慈竹(Dendrocalamus farinosus)是西南地区一个重要的丛生经济竹种,具有抗寒、耐瘠薄、纤维素含量高、材性好等特点,是制浆造纸的优良原料[4]。以往对梁山慈竹的研究主要集中在生长特性[5]、理化特性与纤维形态[6]、细胞壁机械特性[7]等方面,而由于缺乏梁山慈竹生长发育以及品质特性等相关分子生物学基础研究,通过基因工程手段培育优质高产的梁山慈竹研究进展缓慢。基于转录组高通量测序技术,本试验获得并构建了梁山慈竹实生植株以及体细胞突变体No.30和No.66[8]的转录组unigenes库,并基于梁山慈竹体细胞突变体No.30纤维素和木质素含量均比实生植株高的特点[9],筛选出与纤维素和木质素合成相关差异表达基因,为今后梁山慈竹优质性状相关基因的克隆以及功能分析等研究奠定理论基础。
1 材料和方法
1.1 植物材料
梁山慈竹unigenes库的构建选用3份材料:梁山慈竹成熟种胚离体诱导再生植株No.30、No.66以及同期生长的梁山慈竹实生植株(CK),2013年9月于西南科技大学生命科学与工程学院资源圃分别采集No.30、No.66和CK同期生长30 d、高度一致的梁山慈竹竹笋,并分成上、中、下3个部位,每个部位分别剪碎并充分混合,然后每个部位均取等量样品并充分混匀,液氮速冻后保存于-80℃备用。
1.2 试验方法
1.2.1 总RNA提取 参照RNAprep pure Plant Kit(TIANGEN BIOTECH公司)说明书上的方法提取竹笋总RNA,琼脂糖凝胶电泳及紫外分光光度计用于检测RNA质量。
1.2.2 测序文库的构建及转录组测序 提取的竹笋总RNA经NEBNext Poly(A)mRNA Magnetic Isolation Module(NEB,7490)富集mRNA,缓冲剂(Fragmentation)对RNA片段化处理。以mRNA为模板,采用随机引物法,用NEBNext mRNA Library Prep Master Mix Set for Illum ina(NEB,E6110)和NEBNext Multiplex Oligos for Illum ina(NEB,E7500)构建转录组测序文库。构建好的文库用1.8%琼脂糖凝胶电泳检测文库插入片段大小,然后用Library Quantification Kit-Illum ina GA Universal(Kapa,KK4824)进行QPCR定量。检测合格的文库在Illumina cbot上进行簇的生成,最后用Illumina HiSeqTM2000进行测序。
1.2.3 测序数据的组装及差异表达基因筛选 转录组测序得到的原始序列经过去除杂质和冗余处理后,利用Trinity[10]软件对经过过滤后的高质量数据进行de novo拼接,得到各个样本的Contig,随后根据Contig结果,利用paired-end信息做进一步的序列拼接,得到各个样本的转录本Transcript,在转录本聚类单元中选取最主要的Transcript作为各个样本的unigenes序列。
差异表达基因的筛选是建立在同一套参考基因的基础上,对各样本得到的unigenes数据通过cd-hit聚类去冗余,采用TGICL的聚类组装策略最终得到非冗余的梁山慈竹笋期All-unigenes数据库,并通过RPKM[11](Reads per kilobase per million mapped reads)计算样本间的基因表达差异。差异表达基因的筛选条件:错误发现率(False discovery rate)<0.01且RPKM值的倍数变化(Fold Change)在2倍以上。
1.2.4 功能注释、分类及代谢途径分析 利用Blast软件(E-value<1e-05)将梁山慈竹All-unigenes序列及差异表达基因序列分别与Nonredundant protein(Nr)、UniProt/Swiss-Prot、Kyoto Encyclopedia of Genes and Genomes(KEGG)、Cluster of Orthologous Groups of proteins(COG)和Gene Ontology(GO)数据库进行序列比对,获得梁山慈竹All-unigenes及差异表达基因的功能注释信息。
2 结果与分析
2.1 Illum ina H iSeqTM 2000测序和序列拼接
提取笋RNA通过Illum ina平台测序,共得到86 575 631条reads,总碱基数为17.48 Gb。Q30值均达到80.00%以上(表1),可见,本次测序量与测序质量为后续的数据组装提供很好的原始数据。CK以及No.30和No.66经高通量测序,整合得到梁山慈竹unigenes 84 741条,总长度为72.70 Mb, ?平均长度约为857.89 bp,N50长度为1 595 bp。长度为200~300 bp的unigenes所占比例最大,为31.86%;长度大于1 kb的unigenes有23 111条;所占比例为27.27%(表2)。
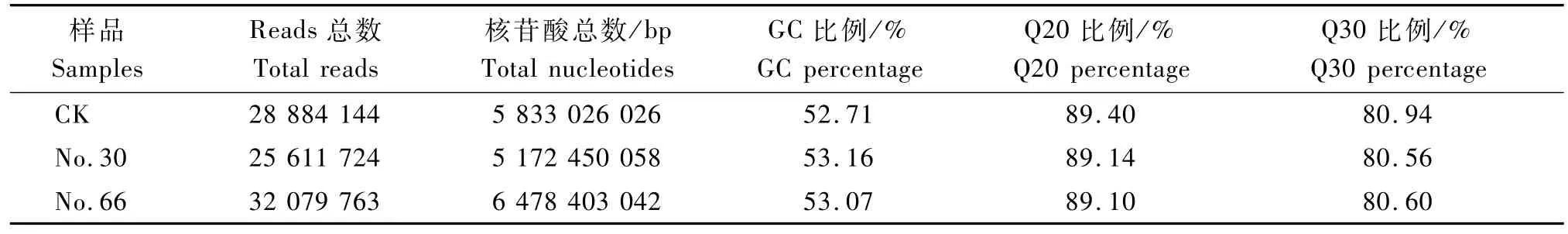
表1 样品测序数据统计Tab.1 Summ ary of Sam p le sequencing data
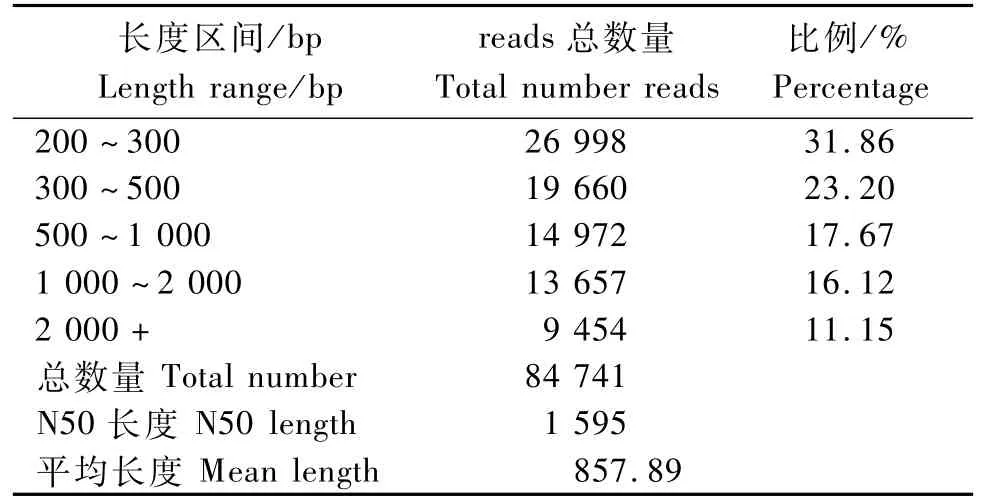
表2 梁山慈竹A ll-unigenes长度分布Tab.2 Distribution of A ll-unigenes length of Dendrocalamus farinosus
2.2 梁山慈竹转录物A ll-unigenes的功能注释
为从整体上了解转录物All-unigenes序列功能信息,对拼接组装的转录物进行Nr、Swiss-Prot、KEGG、COG和GO数据库比对、注释。通过NCBI的BlastX比对,有49 688条转录物被注释到Nr数据库,36 907条转录物被注释到Swiss-Prot数据库(表3)。84 741条转录物中共有49 829条得到注释。
2.3 差异表达基因的分析
2.3.1 差异表达基因的筛选 梁山慈竹体细胞突变体No.30纤维素和木质素含量均明显高于CK,基于CK与No.30这2个样本的转录组测序数据筛选纤维素和木质素生物合成相关差异表达基因。差异表达基因的筛选与分析有助于初步了解No.30高纤维素和木质素含量产生的机制并能为相关基因克隆提供重要基因序列信息。从2个样本unigenes库中共有3 572条差异表达unigenes被筛选出来,2 655条被注释到Nr数据库,2 062条被注释到Swiss-Prot数据库(表4)。
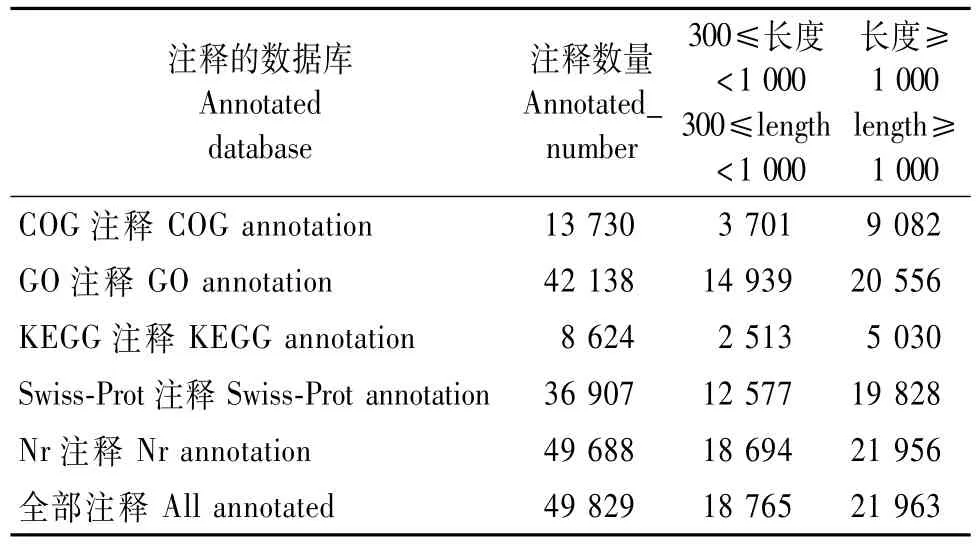
表3 梁山慈竹A ll-unigenes功能注释Tab.3 A ll-unigenes function annotation of Dendrocalamus farinosus

表4 注释的差异基因数目统计Tab.4 Summ ary of differen tial expression gene annotated
2.3.2 差异表达基因的COG分析 COG数据库的目的是对基因产物进行直系同源分类。在COG分类体系中,757条差异表达unigenes具有详细的蛋白功能释义,共获得1 086个COG注释,涉及细胞结构、信号转导、次生代谢等25个COG功能分类。一般功能注释(General function prediction only)代表最大的一类,所占比例为25.10%;其次复制、重组、修复(Replication,recombination and repair)所占比例为17.17%(图1)。此外,该转录组还主要涉及转录(Transcription)、信号转导机制(Signal transduction mechanisms)、碳水化合物的运输和代谢(Carbohydrate transport and metabolism)、氨基酸转运和代谢(Amino acid transport and metabolism)等功能定义。
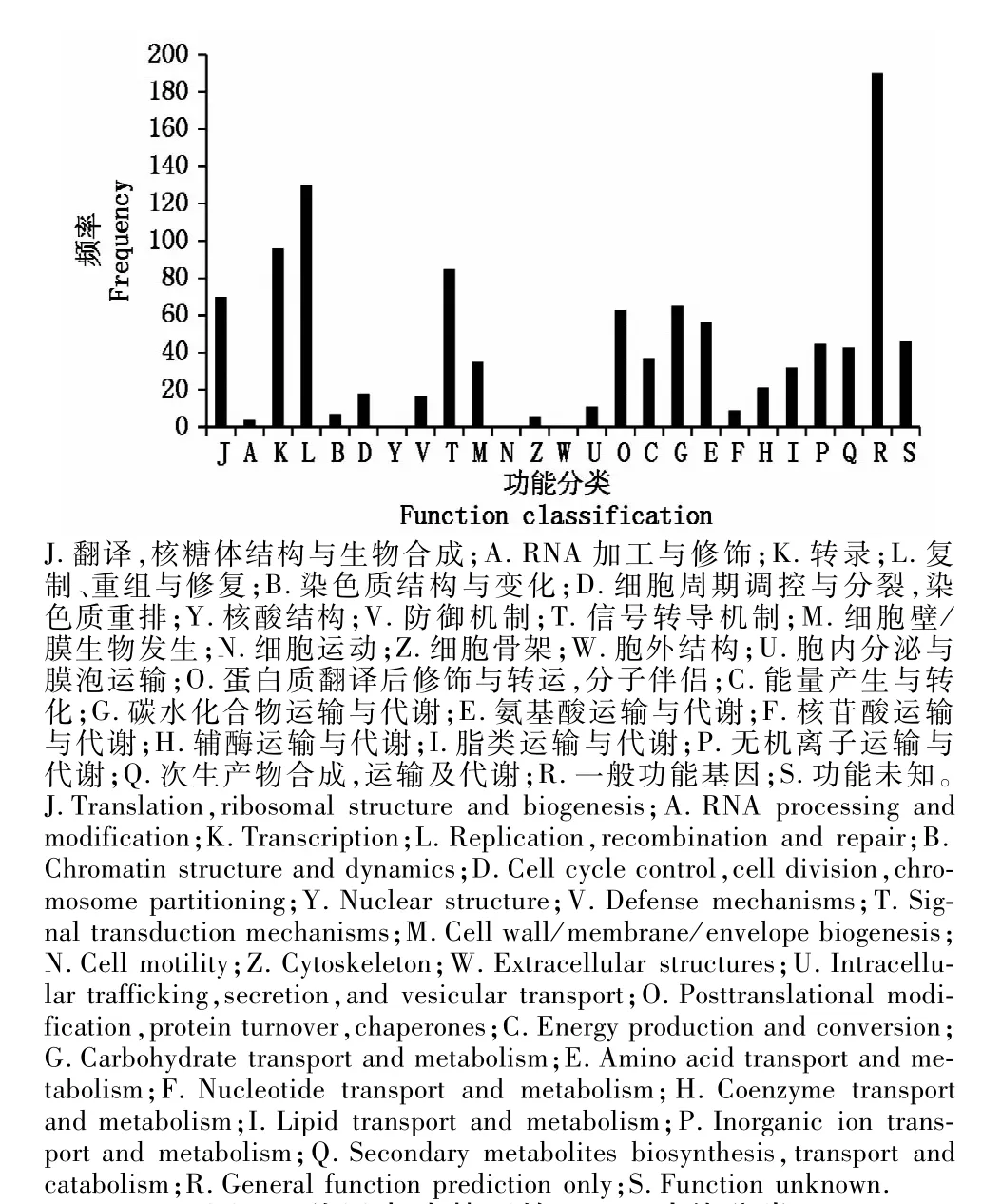
图1 差异表达基因的COG功能分类Fig.1 Differential expression gene COG function classification
2.3.3 差异表达基因的GO分析 为进一步了解差异表达基因的功能,筛选得到序列注释到GO数据库,有2 213条差异表达unigenes具有功能定义,分别注释到细胞组分(Cellular component)、分子功能(Molecular function)、生物过程(Biological process)3个大的功能类别,而上述3大功能类别又可以被划分为更详细的62个亚类,分别包含了18,18,26个功能亚类(图2)。在细胞组分功能类型中,细胞(Cell)和细胞部分(Cell part)2个功能亚类所占比例最高;在分子功能类型中,结合(Binding)和催化活性(Catalytic activity)2个功能亚类所占比例最高;在生物过程功能类型中,细胞过程(Cellular process)和代谢过程(Metabolic process)所占比例最高。
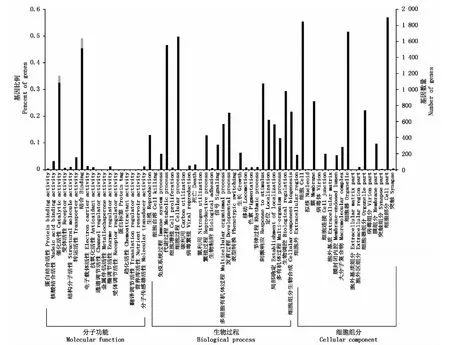
图2 差异表达基因的GO功能注释Fig.2 Differential expression gene GO function annotation
2.3.4 差异表达基因KEGG Pathway功能注释基因间的相互作用对于生物体行使生物学功能有着非常重要的作用,为了鉴定在代谢或者信号通路中显著富集的基因,将差异表达基因映射到KEGG数据库,得到385个功能定义,被注释到94条KEGG Pathways中。差异表达基因注释序列最多的10条代谢通路为:嘌呤代谢(Purine metabolism)、光合有机体碳固定(Carbon fixation in photosynthetic organisms)、半胱氨酸和甲硫氨酸代谢(Cysteine and methionine metabolism)、苯丙烷类生物合成(Phenylpropanoid biosynthesis)等(表5)。

表5 差异表达基因KEGG功能注释Tab.5 Differential expression genes KEGG function annotation
2.3.5 纤维素和木质素合成相关差异表达基因分析 依据差异表达基因在Swiss-Prot蛋白数据库功能释义,并根据RPKM值大小对纤维素和木质素生物合成相关差异表达基因进行分析,结果表明,纤维素合酶、过氧化物酶、泛素连接酶和热休克蛋白在梁山慈竹体细胞突变体No.30中表达量升高,MYB4、4-香豆酸CoA连接酶、肉桂醇脱氢酶、肉桂酰-CoA还原酶和漆酶在突变体中表达量降低(表6)。
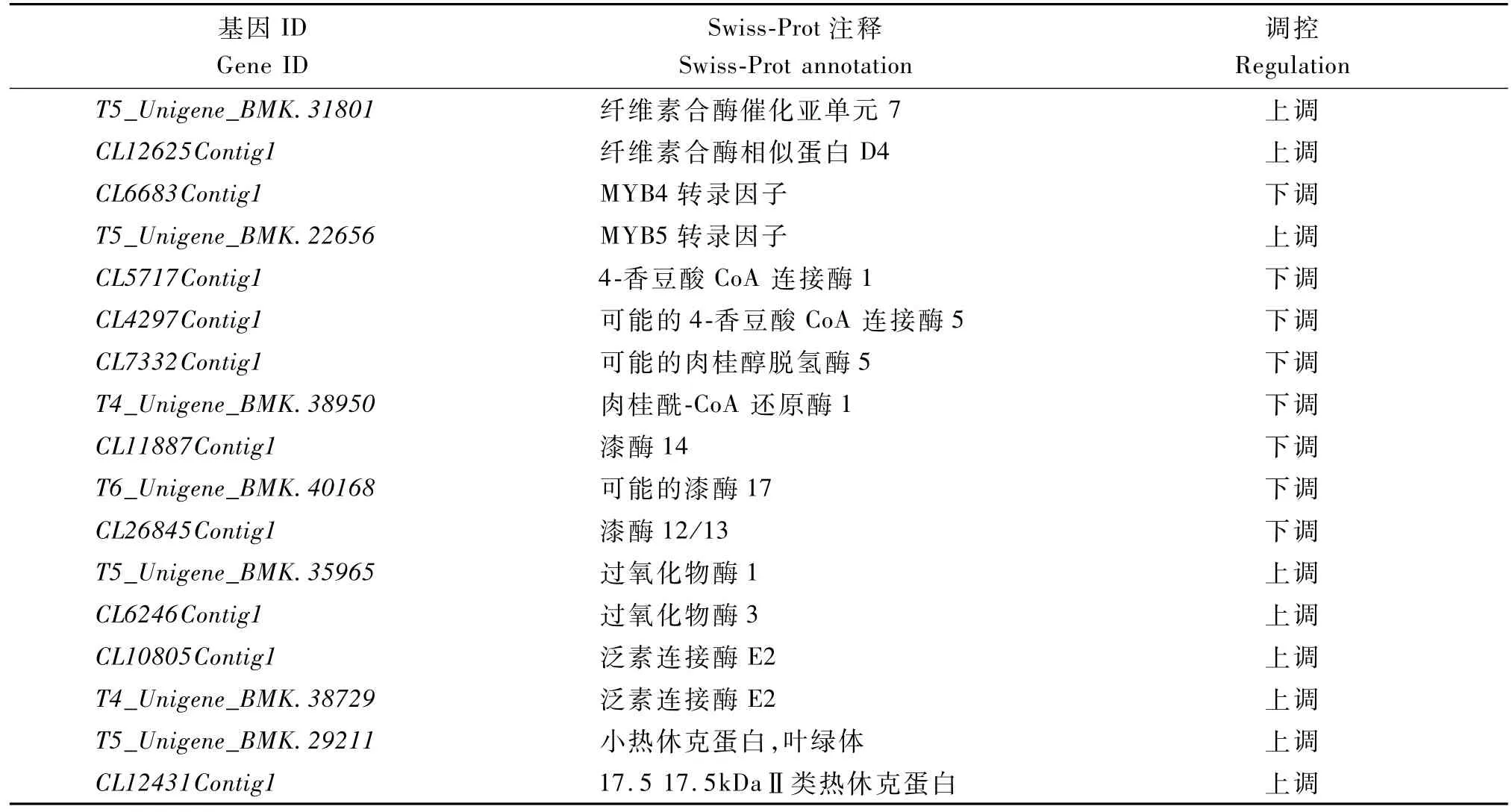
表6 纤维素和木质素生物合成相关差异表达基因Tab.6 Differential expression gene related to cellulose and lignin biosynthesis
3 讨论
伴随着测序技术的发展,拟南芥[12]、水稻[13]、毛竹[14]等基因组得到测序,但是由于价格昂贵,大多数植物的基因组并不能得到测序。高通量测序技术的出现对于挖掘基因以及加深非模式植物生长和发育的理解提供了新方法。为尽可能完整获得梁山慈竹转录组信息,本试验选用了包括梁山慈竹实生植株及其体细胞突变体No.30、No.66在内的3份梁山慈竹转录组样本进行了转录组测序。测序共获得17.48 Gb数据量,reads数86 575 631条,de novo组装后获得84 741条unigenes,其中长度在1 kb以上unigenes有23 111条。这些结果表明,本研究所采用的双端测序(paired-end)的方法,增加了测序深度,同时也提高了de novo拼接的效率和准确性[15]。
梁山慈竹All-unigenes以及差异表达基因在Nr以及GO等数据库的注释信息,为基因组数据严重匮乏的梁山慈竹开展功能基因组学研究提供了新的思路和方法。KEGG注释结果显示,差异表达基因富集的通路主要有碳的固定、苯丙烷类生物合成等生物化学途径,这些途径与纤维素和木质素生物合成有紧密的联系。根据Swiss-Prot蛋白数据库功能释义,从梁山慈竹体细胞突变体No.30植株中发现2个具有差异表达的MYB转录因子,部分MYB类转录因子通常结合到木质素合成基因启动子区域的AC元件调控苯丙烷代谢途径[16]。有研究表明,MYB4对苯丙烷代谢途径表现出不同的调控效应[17-18],本试验中梁山慈竹体细胞突变体No.30中MYB4的下调表达对苯丙烷代谢途径的调控机制需进一步研究。MYB5则被认为主要参与到种皮的发育过程[19]。在4-香豆酸CoA连接酶、肉桂醇脱氢酶和肉桂酰-CoA还原酶等木质素生物合成相关基因表达下调的同时,推测过氧化物酶上调表达是其木质素含量升高的重要原因。纤维素合酶7在纤维素合成过程中发挥重要作用[20],梁山慈竹体细胞突变体No.30中纤维素合酶7的上调表达推测是其纤维素含量升高的重要原因。热休克蛋白以及泛素连接酶在细胞伸长过程中能有效控制过氧化氢以及活性氧的含量[21],这2种基因在突变体中表达量的升高可能赋予其更好的纤维特性。以上相关蛋白功能还有待进一步克隆验证。
植物组织培养是植物科学研究的一个重要手段,广泛用于提高植物抗胁迫能力[22]、珍稀濒危物种保护[23]、代谢产物产量提高[24]等多个方面。以往对于体细胞变异的研究主要采用RAPD、形态观察、染色体计数等方法[25-27],但是对于基因在转录水平上的变化一无所知;此外,竹子作为一种非木材类造纸资源,其纤维素含量、纤维质量以及木质素含量成为关注重点,RNA高通量测序技术在梁山慈竹及其体细胞突变体No.30上的应用以及纤维素、木质素生物合成相关基因的筛选为竹类遗传改良研究提供了珍贵的资源。
[1] 刘红亮,郑丽明,刘青青,等.非模式生物转录组研究[J].遗传,2013,35(8):955-970.
[2] Li X,Sun H,Pei J,et al.De novo sequencing and comparative analysis of the blueberry transcriptome to discover putative genes related to antioxidants[J].Gene,2012,511(1):54-61.
[3] Liu M,Qiao G,Jiang J,et al.Transcriptome sequencing and de novo analysis for Ma bamboo(Dendrocalamus latiflorus Munro)using the Illumina platform[J].PLoS One,2012,7(10):e46766.
[4] 熊 壮,扶志宏,鄢武先,等.四川梁山慈竹纸浆林生产现状及培育技术初步研究[J].世界竹藤通讯,2012,10(5):7-11.
[5] 李永全,张安刚,龚兆全,等.梁山慈竹特性及育苗技术[J].四川林勘设计,2012(2):34-37.
[6] 胡尚连,蒋 瑶,陈其兵,等.四川2种丛生竹理化特性及纤维形态研究[J].植物研究,2010,30(6):708-712.
[7] Yang X,Tian G L,Shang L L,et al.Variation in the cell wall mechanical properties of Dendrocalamus farinosus bamboo by nanoindentation[J].BioResources,2014,9(2):2289-2298.
[8] Hu S L,Jy Z,Cao Y,et al.In vitro callus induction and plant regeneration from mature seed embryo and young shoots in a giant sympodial bamboo,Dendrocalamus farinosus(Keng et Keng f.)Chia et HL Fung[J].A frican Journal of Biotechnology,2011,10(16):3120-3125.
[9] 郭鹏飞.梁山慈竹体细胞无性系再生植株变异研究[D].绵阳:西南科技大学,2013.
[10] Grabherr M G,Haas B J,Yassour M,et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J].Nature Biotechnology,2011,29(7):644-652.
[11] Mortazavi A,W illiams B A,Mccue K,et al.Mapping and quantifying mammalian transcriptomes by RNA-Seq[J]. Nature Methods,2008,5(7):621-628.
[12] Kaul S,Koo H L,Jenkins J,et al.Analysis of the genome sequence of the flowering plant Arabidopsis thaliana[J]. Nature,2000,408(6814):796-815.
[13] Goff S A,Ricke D,Lan T H,et al.A draft sequence of the rice genome(Oryza sativa L.ssp.japonica)[J]. Science,2002,296(5565):92-100.
[14] Peng Z,Lu Y,Li L,et al.The draft genome of the fastgrowing non-timber forest species moso bamboo(Phyllostachys heterocycla)[J].Nature Genetics,2013,45(4):456-461,461e1-2.
[15] Fullwood M J,Wei C L,Liu E T,et al.Next-generation DNA sequencing of paired-end tags(PET)for transcriptome and genome analyses[J].Genome Research,2009,19(4):521-532.
[16] Li C F,Wang X Q,Lu W X,et al.A pop lar R2R3-MYB transcription factor,PtrMYB152,is involved in regulation of lignin biosynthesis during secondary cell wall formation[J].Plant Cell,Tissue and Organ Culture,2014,119(3):553-563.
[17] Patzlaff A,Mcinnis S,Courtenay A,et al.Characterisati-on of a pine MYB that regulates lignification[J].The Plant Journal,2003,36(6):743-754.
[18] Ma Q H,Wang C,Zhu H H.TaMYB4 cloned from wheat regulates lignin biosynthesis through negatively controlling the transcripts of both cinnamyl alcohol dehydrogenase and cinnamoyl-CoA reductase genes[J].Biochimie,2011,93(7):1179-1186.
[19] Li S F,M illiken O N,Pham H,et al.The Arabidopsis MYB5 transcription factor regulates mucilage synthesis,seed coat development,and trichome morphogenesis[J].The Plant Cell,2009,21(1):72-89.
[20] Taylor N G,Laurie S,Turner SR.Multiple cellulose synthase catalytic subunits are required for cellulose synthesis in Arabidopsis[J].The Plant Cell,2000,12(12):2529-2540.
[21] Hovav R,Udall JA,Chaudhary B,et al.The evolution of spinnable cotton fiber entailed prolonged development and a novel metabolism[J].PLOS Genetics,2008,4(2):e25.
[22] Sengar A S,Thind K S,Kumar B,et al.In vitro selection at cellular level for red rot resistance in sugarcane(Saccharum sp.)[J].Plant Growth Regulation,2009,58(2):201-209.
[23] 梁 芳,蒋素华,王洁琼,等.濒危植物太行菊组织培养及快繁技术研究[J].中国农学通报,2015,31(16):115-120.
[24] 王鹏姬.荞麦愈伤组织培养及其黄酮合成研究[D].杨凌:西北农林科技大学,2013.
[25] Sheidai M,Aminpoor H,Noormohammadi Z,et al.RAPD analysis of somaclonal variation in banana(Musaacuminate L.)cultivar Valery[J].Acta Biol Szeged,2008,52:307-311.
[26] Carsono N,Yoshida T.Variation in spikelet-related traits of rice plants regenerated from mature seed-derived callus culture[J].Plant Production Science,2007,10(1):86-90.
[27] Mohanty S,Panda M K,Subudhi E,et al.Plant regeneration from callus culture of Curcuma aromatica and in vitro detection of somaclonal variation through cytophotometric analysis[J].Biologia Plantarum,2008,52(4):783-786.
H igh-throughput RNA-seq and Analysis on Differential Expressed Gene from Dendrocalamus farinosus
WANG Shenchang1,2,HU Shanglian1,2,CAO Ying1,2,XU Gang1,2
(1.Lab of Plant Cell Engineering,Southwest University of Science and Technology,Mianyang 621010,China;2.Engineering Research Center for Biomass Resource Utilization and Modification of Sichuan Province,Mianyang 621010,China)
The transcriptome from the shoots of Dendrocalamus farinosus and its somatic mutantwas sequenced using the RNA-Seq technology to elucidate its functional gene and analyze its differential expressed gene,providing a theoretical basis for its genetic improvement.The sequencing data was assembled by de novo assembly and the differential expressed gene screened was annotated in COG,GO and KEGG database.In addition,the differential expression of gene related to cellulose and lignin biosynthesis was analyzed,basing on the Swiss-Prot function annotation.The sequencing results showed that a total of 86 575 631 reads were produced and assembled into a total of 84 741 unigenes by de novo,among which 49 829 were annotated in Nonredundant protein,Cluster of Orthologous Groups of proteins,Gene Ontology,Kyoto Encyclopedia of Genes and Genomes and Swiss-Prot database.Besides,a total of 3 572 differential expressed unigenes were identified from the plant of seeds(CK)and the somatic mutant No.30.Of these differential expressed genes,757 unigenes had a detailed protein functions in the COG classification system,2 213 unigenes had the function definition in the GO database,385 unigenes were annotated to 94 KEGG Pathways.The expression of genes encoding CesA,Prx,Ubiquitin-conjugating enzyme and Heat shock proteins increased in somatic mutant No.30,while,the expression of genes encoding MYB4,4CL,CAD,CCR and LAC decreased.Our data provide themost comprehensive transcriptom ic resource for Dendrocalamus farinosus and the tran-script sequences with important function related to cellulose and lignin biosynthesis were found,which provide the most precious information resources for the further research on bamboo.
Dendrocalamus farinosus;Somatic mutant;Transcriptome;Differential expressed gene
Q78 文献标识码:A 文章编号:1000-7091(2016)03-0065-07
10.7668/hbnxb.2016.03.010
2016-03-10
国家自然科学基金青年基金项目(31400257;31400333);四川省“十三五”育种公关资助项目;四川省生物质资源利用与改性工程技术研究中心基金项目(12zxsk07;13zxsk01);西南科技大学研究生创新基金项目(15ycx092)
王身昌(1989-),男,山东菏泽人,在读硕士,主要从事植物遗传与品种改良研究。
胡尚连(1966-),女,河北秦皇岛人,教授,博士,主要从事植物生理与生物技术研究。
猜你喜欢
趣味(作文与阅读)(2022年5期)2022-07-29
戏曲研究(2021年4期)2021-06-05
今日农业(2020年24期)2020-12-15
科学(2020年4期)2020-11-26
岷峨诗稿(2019年4期)2019-04-20
专用汽车(2018年10期)2018-11-02
生命科学研究(2018年1期)2018-05-29
上海农业学报(2017年3期)2017-04-10
安徽医科大学学报(2016年12期)2017-01-15
中西医结合心脑血管病杂志(2016年20期)2016-03-01
