
基于极端混合池(BSA)全基因组重测序的甘蓝型油菜 有限花序基因定位
2018-09-03 11:03张尧锋张冬青余华胜林宝刚华水金丁厚栋傅鹰
中国农业科学 2018年16期
张尧锋,张冬青,余华胜,林宝刚,华水金,丁厚栋,傅鹰
基于极端混合池(BSA)全基因组重测序的甘蓝型油菜 有限花序基因定位
张尧锋,张冬青,余华胜,林宝刚,华水金,丁厚栋,傅鹰
(浙江省农业科学院作物与核技术利用研究所,杭州 310021)
【目的】适合机械化收获是当今油菜育种改良和遗传研究的重要目标。该研究以一个自然变异产生的油菜有限花序(,)突变体为研究对象,通过分析有限花序的遗传模式,开展有限花序性状的基因定位和克隆,以期发掘候选基因,为培育适合机械化收获的油菜新品种提供新思路和新材料,为揭示油菜有限花序遗传机制奠定基础。【方法】以一个稳定遗传的有限花序突变株系FM8与野生型自交系FM7开展正反交,观察F1和F2后代的花序形态,分析有限花序性状的遗传模式。在F2群体中挑选20个有限花序单株和20个野生类型单株构建混合池,对混合池和亲本开展20×和10×覆盖度的全基因组重测序,定位有限花序性状的关联区间。根据关联区间对应到拟南芥基因组的共线性区段和基因注释信息,预测候选基因,并对候选基因进行同源克隆,发掘序列变异,筛选关键基因。【结果】油菜有限花序突变性状表现为初花期主花序和侧枝花序顶部形成一个或若干个顶生花,花序无限生长受阻,导致结角期主枝和侧枝有封顶特征即有限花序。有限花序突变株系与野生型正反交F1均表现为野生型,F2代无限花序与有限花序的分离比符合13﹕3,说明有限花序的遗传受2对隐性基因和1对隐性上位抑制基因互作控制。对混合池及亲本开展全基因组重测序,得到30 123个单核苷酸多态性(SNPs)标记和107 636个插入缺失标记(InDels)标记,用于有限花序性状的全基因组定位。定位结果共检测获得7个显著关联区间,分布于油菜A08、A09、A10、C08和C09共5条染色体。其中,A10染色体上的关联区间峰值最高,是控制有限花序性状的主效位点。并且,A10染色体关联区间内的14.36—15.07 Mb的区域与C09染色体2个关联区间显示高度同源性。候选基因预测发现位于A08、A09、A10、C08和C09的5个关联区间包含有8个候选基因,包括()、()、()、()和()。基因序列分析表明突变体、和的基因编码区存在序列变异,并导致蛋白序列变异。【结论】油菜有限花序突变由2对隐性基因和1对隐性上位抑制基因互作控制。与有限花序性状显著关联的区间有7个,其中,位于染色体A10和C09的关联区间具有高度同源性。、和被推断为有限花序性状的候选基因。
甘蓝型油菜;有限花序;基因定位;关联区间;候选基因
0 引言
【研究意义】中国是世界油菜生产大国,种植面积7×106hm2,总产1.1×107t,均占全世界的三分之一(www.fao.org)。油菜生产在中国农业中有着举足轻重的作用。然而,随着中国农村劳动力成本增加,油菜种植效益和种植面积逐年下降[1],急需筛选和培育具有早熟、矮秆、株型紧凑、分枝短、熟期整齐度好等特性的品种,以适应油菜机械化生产的需要。由于油菜驯化历史短,变异类型有限,很大程度限制了油菜的遗传改良。因此,发现和创制新的油菜变异类型对拓宽现有油菜遗传资源、加快选育适合机械化作业的油菜品种有重要意义。【前人研究进展】高等植物的花序分限花序和无限花序[2]。自然生长的(野生型)油菜属于无限花序类型,即随着花序轴的伸长,不断产生花芽、形成花蕾。具有无限生长习性的油菜在生产上存在一些缺陷,如:植株较高、易倒伏;结角层不集中,整齐度不好;熟期不一致,无效角果多[3]。实现油菜从无限花序向有限花序转变,克服无限花序生长习性的不足,是选育适合机械化收获的油菜品种的新思路。高等植物开花现象涉及一系列精确的发育进程[4-10]。如果植株花序分生组织表现持续激活,就会源源不断产生花分生组织,表现为无限花序类型。目前,在一些物种中已发现无限花序植株的有限花序变异。Shannon等[11]通过EMS诱变产生了有限花序类型的拟南芥;Bradley等[12]发现了一个自然变异的金鱼草有限花序突变体;Dhanasekar等[13]通过γ射线诱导豌豆产生了有限花序突变体;Kaur等[14]在人工合成芥菜型油菜创制过程中产生了有限花序变异类型的芥菜型油菜;Zhang等[15]通过EMS诱变产生了芝麻有限花序突变体;刘毅[16]发现了自然突变的有限花序芝麻突变体。在不同物种的有限花序突变体中,花序形态基因突变被认为是控制无限花序向有限花序变异的关键因素。例如,金鱼草的基因()是最早克隆到的导致无限花序向有限花序变异的花序形态基因[12]。在随后的研究中发现,不同物种的直系同源基因也与植株从无限花序向有限花序变异相关,包括拟南芥()[17]、番茄()[18]、烟草()[19]、黑麦草[20]和豌豆[21],近年来报道的柑橘[22]、水稻[23]、苹果[24]等物种中的/同源基因均被认为是控制花序变异的关键基因。在一些物种的有限花序突变体中,已经定位了基因所在的关键区间:如在芝麻中,有限花序调控基因定位到连锁群LG8上18.0—19.2 cM的遗传区间[15];芸薹属人工合成芥菜型油菜中的有限花序突变体[25-26],其突变基因定位到B5染色体上;甘蓝型油菜中,通过小孢子培养得到的一个有限花序双单倍体(DH)株系,经遗传分析发现有限花序的遗传模式为隐性单基因调控,其调控基因定位到A10染色体68 kb的物理区段,预测为关键候选基因[27]。【本研究切入点】尽管目前已经在若干无限花序物种中发现了有限花序突变体,并且相关基因已经被定位甚至克隆,但在油菜上关于有限花序突变体及相关遗传机制的研究鲜见报道[27]。【拟解决的关键问题】本研究发现了一个与前人报道不同的自然产生的油菜有限花序突变体(,),基于高通量测序平台对突变体和野生型以及F2代的2个极端混合池开展全基因组重测序,定位与油菜有限花序性状显著关联的区域,并通过基因克隆测序筛选关键候选基因,为适合机械化收获的油菜新品种培育提供新思路和新材料,同时也为揭示油菜有限花序遗传机制奠定基础。
1 材料与方法
1.1 材料
2008年在育种材料中发现了一株油菜有限花序自然突变体,连续5年套袋自交获得稳定遗传的突变株系FM8。2015年以突变株系FM8与正常自交系FM7正反交获得F1和F2。从FM8为母本与FM7杂交构建F2分离群体中挑选20个有限花序性状明显的F2单株和20个正常F2单株构建成突变类型混合池和野生类型混合池,用于有限花序性状基因定位。
1.2 田间试验和性状调查
经过多年田间观察,确定油菜有限花序性状的评价方法如下:花期花序生长轴顶端发育形成顶生花,花序继续生长和后续花蕾形成均受阻,结角期表现出主枝和侧枝花序顶端没有出现无效角果,有封顶的特征。
2016年花期,F1单株套袋,收获自交种子获得F2。2016年10月5日采用穴盘育苗方式播种F2以及2个亲本种子,11月10日移栽到浙江省农业科学院试验大田。田间种植方式为3行区,行距为0.4 m,株距为0.3 m。田间管理按照当地常规方式进行,水平一致。初花期观察每个F2单株花序生长情况,并挂牌记录,终花期再次观察确认F2单株的花序状态。
1.3 极端混合池构建与基因分型
F2群体中挑选20株有限花序性状明显的突变单株和20株野生类型单株,连同2个亲本FM8和FM7,取幼嫩植株叶片0.15 g,利用OMEGA HP Plant DNA提取试剂盒提取DNA。将20个有限花序F2单株和20个野生型F2单株的DNA分别等量混合,构建成突变性状混合池和野生类型混合池。2个混合池DNA和2个亲本DNA按照标准流程构建文库,并通过illumina HiSeqTMPE150对2个混合池和两亲本开展20×和10×覆盖度的全基因组重测序。测序得到的原始序列去除接头和低质量序列,再通过BWA软件[28](参数:mem-t4-k32-M)比对到油菜参考基因组(http://www. genoscope.cns.fr/brassicanapus/data/Brassica_napus_v4.1. chromosomes.fa.gz)。比对结果经SAMTOOLS的rmdup命令去除重复[29]。采用GATK3.3软件的UnifiedGenotyper模块进行多个样本单核苷酸多态性(SNPs)标记和插入缺失(InDels)标记的检测,使用VariantFiltration进行过滤[30],并利用ANNOVAR软件对SNP和InDel进行注释[31]。
1.4 基于混合池的有限花序性状基因定位与候选基因预测
基于基因分型的结果,筛选亲本间纯合差异的多态性位点。选择亲本FM8作为参照亲本,参考Takagi等[32]的方法计算2个子代混合池在每个多态性位点上的SNP频率(SNP-index)。为减少测序错误和比对错误造成的影响,对计算出SNP-index后的亲本多态性位点进行过滤,过滤标准如下:(1)2个子代中SNP-index都小于0.3,并且SNP深度都小于7的位点,过滤掉;(2)1个子代SNPindex缺失的位点,过滤掉。随后,计算2个子代SNP-index作差(△SNP-index)。选择1 Mb为窗口、1 kb为步长对△SNP-index在各个染色体上的分布进行作图,选取95%置信水平作为筛选的阈值,置信水平以上的窗口作为候选区间。为了预测关联区间内的候选基因,我们收集了拟南芥中与花序和花发育相关的基因(数据来源于BRAD数据库http://brassicadb.org/brad/),利用blast2go[33]中的blastall将这些基因的CDS序列与101 040个油菜基因的CDS序列进行比对,以比对覆盖率(>70%)和值(<1e-50)为标准寻找对应到油菜上的同源基因。如果花序和花发育相关基因对应到油菜上的同源基因位于显著关联的候选区间,则认为该基因为候选基因。
1.5 候选基因分子克隆和序列分析
参照油菜基因组数据库(http:// brassicadb. org/brad/)公布的目的基因序列设计引物(电子附表1)。以有限花序和无限花序亲本,以及有限花序和无限花序的F2株系的基因组DNA为模板进行扩增,电泳后回收纯化,连接、转化,PCR检测,挑选出与预测片段分子量大小一致的阳性克隆送到公司测序。
2 结果
2.1 油菜有限花序性状的形态观察与鉴定
油菜有限花序突变体在初花期表现出主花序和侧枝花序顶部形成1个或者若干个柱头外露的顶生花(图1-A),此类型顶生花不仅自身无法正常开花,同时也限制了花序生长和后续花蕾形成。结角期,突变体表现出主枝和侧枝有封顶的特征(图1-C)。由于存在营养竞争,无限花序顶端角果结实率往往显著降低,产生无效角果(图1-D)。而有限花序顶部通常不存在无效角果(图1-C)。由于有限花序的形成,突变体表现出株高降低、成熟期更一致并且熟期提早等有利特征。除此之外,有限花序突变体与野生型油菜的其他农艺性状并无明显差别,表现为植株粗壮,叶色较深,分裂较浅,叶面光滑,初花期为3月10日。除顶端花蕾柱头外露,其他部位的花器发育良好,花瓣平滑。

A:花期的油菜有限花序突变体;B:花期的野生型油菜;C:结角期的油菜有限花序突变体;D:结角期的野生型油菜
2.2 油菜有限花序性状的遗传分析
以稳定遗传的油菜有限花序株系FM8作为亲本,与野生型油菜自交系FM7正反交,F1均表现为野生型无限花序,说明该有限花序突变为隐性突变。突变株系FM8与正常自交系FM7构建的F2群体中出现了有限花序与无限花序的分离,卡方测验表明分离比符合13﹕3分离(表1),说明油菜有限花序性状受2对隐性基因和1对隐性上位抑制基因互作控制。
2.3 极端群体混合池构建与测序数据分析
对20个有限花序F2单株和20个无限花序F2单株构建的混合池以及两亲本开展全基因组重测序,总共得到53.17 G的原始数据。过滤后4个样本的有效序列数据量在8 722.79—17 717.90 M,总数据量为53.026 G。测序数据Q20>96.9%,Q30>95.29%,GC含量在37.35%—38.73%,测序数据有95.36%—98.56%的序列可以成功比对到参考基因组。由此可知,所有样本的数据量足够,测序质量合格,GC分布正常,测序数据与油菜参考基因组比对结果正常,可用于后续的变异检测及性状的基因定位。
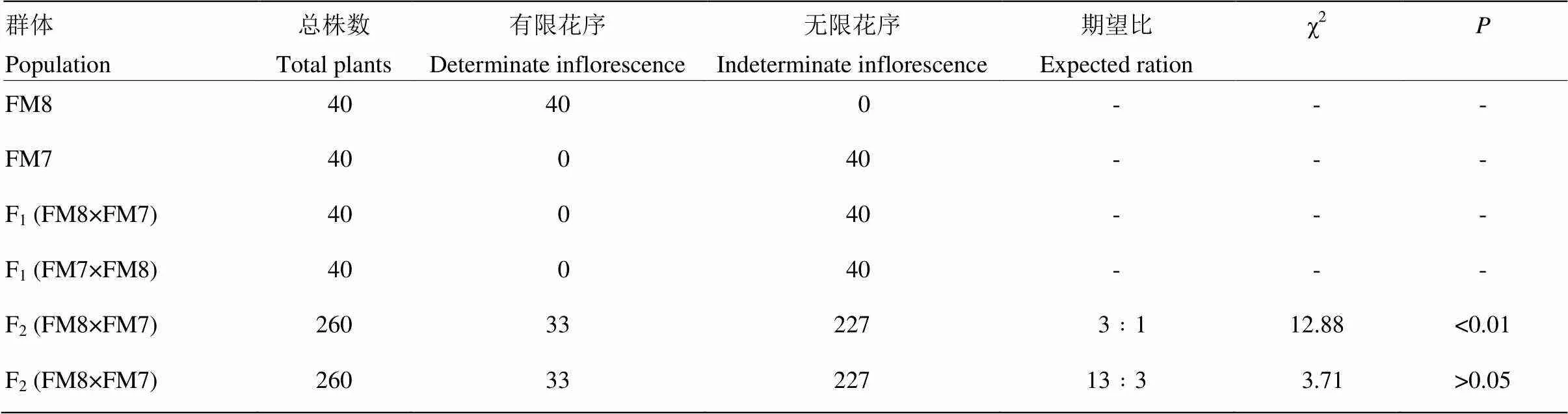
表1 有限花序突变株系与野生型杂交组合后代分离比例
将突变体亲本FM8和野生型亲本FM7测序结果比较,共获得1 186 750个SNPs和764 900个InDels。亲本间检测到的SNPs明显多于InDels。根据变异发生的位置进行比较发现,无论SNPs变异还是InDels变异,位于基因非编码区的多态性位点显著多于基因编码区。基于基因分型的结果,筛选2个亲本间纯合差异的标记,最终挑选出137 759个多态性位点,包括30 123个SNPs和107 636个InDels,用于后续基因定位。这些多态性位点在染色体间呈现非均匀分布,其中A07染色体拥有最丰富的多态性位点,而A04染色体拥有最少的多态性位点。
2.4 油菜有限花序性状基因定位
分析2个混合池△SNP-index,用于甘蓝型油菜有限花序性状的基因定位。定位结果显示,分布于甘蓝型油菜5条染色体的7个区域出现了超过临界值水平的极显著峰,说明这些区域可能包含调控油菜有限花序变异的基因(图2)。这7个显著关联区间在油菜基因组上的分布为chr.A08:12.36—12.51 Mb、chr.A09:26.01—26.05 Mb、chr.A09:30.34—30.68 Mb、chr.A10:14.43—17.37 Mb、chr.C08:27.38—27.75 Mb、chr.C09:45.80—46.45 Mb和chr.C09:46.54—46.60 Mb。这些显著关联区间覆盖的染色体长度为0.04—3.04 Mb。其中,染色体A10关联区间顶点峰值最高,暗示该区间可能具有引起油菜有限花序变异的主效基因。
2.5 染色体A10主效关联区域与C09关联区域的微共线性比较研究
从表2可以看出,A10染色体关联区间对应到拟南芥基因组上的共线性区段(chr.5:0—4.02 Mb)完全覆盖并延伸了C09染色体2个关联区间对应到拟南芥上的共线性区段(chr.5:3.10—3.57 Mb和chr.5:2.98—3.04 Mb)。进一步分析A10上的关联区间发现,该区间内的14.63—15.07 Mb区段与C09的2个关联区间对应到拟南芥基因组的相同位置(图3),这说明A10染色体14.36—15.07 Mb的关联区域与C09染色体的2个候选区域具有高度同源性。油菜A和C亚基因组上的同源区域同时被检测出与油菜有限花序性状显著连锁不仅证实了这三个候选区间的可信性,并且可以推测这三个区域对油菜有限花序的调控可能由同源遗传因子决定。

表2 有限花序性状7个关联区间对应到拟南芥基因组上的共线性区段
2.6 油菜有限花序性状候选基因预测与克隆
通过数据库收集拟南芥与花序和花发育相关的基因(数据来源于BRAD数据库http://brassicadb.org/ brad/),并比对得到这些基因对应到油菜基因组上的同源基因(电子附表2)。8个基因被鉴定到位于A08、A09、A10、C08和C09的5个关联区间(表3):(,)、()、(,)、()、()、(,)、()和(,)。其中,是花序形态关键基因,被锚定到具有最高峰值的A10染色体主效关联区间,因此,推测为调控有限花序性状的关键基因。

红色曲线代表利用滑动窗口计算得到的整个染色体上ΔSNP-index的变化趋势。绿色虚线代表显著性阈值,如果一个区域的滑动窗口曲线出现明显的峰谷并超过临界值,则认为这个区域与性状显著关联,每个点应对一个SNP,横坐标代表每个SNP在油菜基因组上的物理位置,纵坐标代表ΔSNP-index 值
利用有限花序亲本FM8和有限花序F2株系,野生型亲本FM7和野生型F2株系的基因组DNA对有限花序性状关联区间内的8个潜在候选基因进行基因克隆和测序。结果显示,有3个潜在候选基因发生了编码区的碱基序列变异,并最终导致氨基酸序列变异:位于A10染色体上的主效关联区间内的,有限花序亲本在其编码区存在2个SNPs变异,并最终导致TFL1蛋白序列发生了2个氨基酸的变异(图4);位于A09染色体关联区间内的,有限花序亲本在其编码区存在6个碱基的插入和22个SNPs变异,并最终导致FVE蛋白序列发生了2个氨基酸的插入和8个氨基酸序列变异(图4);位于C08染色体关联区间内的,有限花序亲本在其编码区发生了31个碱基片段的丢失,导致第146—174位置的氨基酸序列发生变异,并且第175位置发生氨基酸终止编码(图4)。
3 讨论
3.1 di1突变体是一个新型的油菜有限花序突变体
在芸薹属作物中已有关于有限花序变异的报道。Kaur等[26]在远缘杂交获得的人工合成芥菜型油菜后代中发现了受隐性单基因控制的芥菜型油菜有限花序突变体。Li等[27]在甘蓝型油菜不同生态型杂交的小孢子培养后代中也发现了受隐性单基因控制的甘蓝型油菜有限花序突变体。与前人报道的芸薹属中的有限花序突变体进行比较发现:(1)从遗传模式上看,本研究中的油菜有限花序突变受2对隐性基因和1对隐性上位抑制基因互作控制。这个遗传模式与前人报道的芸薹属中的有限花序突变体并不完全相同。(2)从突变体产生方式看,上述得到的芥菜型油菜和甘蓝型油菜突变体为远缘杂交或者不同生态型间杂交诱导产生,而本研究中的油菜有限花序突变体为自然突变产生。(3)从基因定位结果看,芥菜型油菜有限花序调控基因定位在B5染色体1.9 cM的区段,但其调控基因尚未报道。Li等[27]报道的甘蓝型油菜有限花序调控基因定位于A10染色体,预测为候选基因,该基因与本研究预测得到的位于主效区域的候选基因相同。然而,除此之外,本研究还定位得到其他调控位点,这与该性状受2对隐性基因和1对隐性上位抑制基因互作控制的遗传模式吻合。(4)尽管在本研究和Li等[27]的报道中均为关键候选基因,但变异位点存在差异。Li等[27]报道中蛋白序列的第46和第47位的氨基酸产生变异,而本研究蛋白变异位点位于第37位和第139位。综上所述,本研究得到的油菜有限花序突变体与前人报道的有限花序突变体并不相同,该新型油菜有限花序突变体的发现对于拓宽甘蓝型油菜的变异类型具有一定意义。针对预测得到的候选基因开展更加深入的功能验证将在后续研究中继续开展。
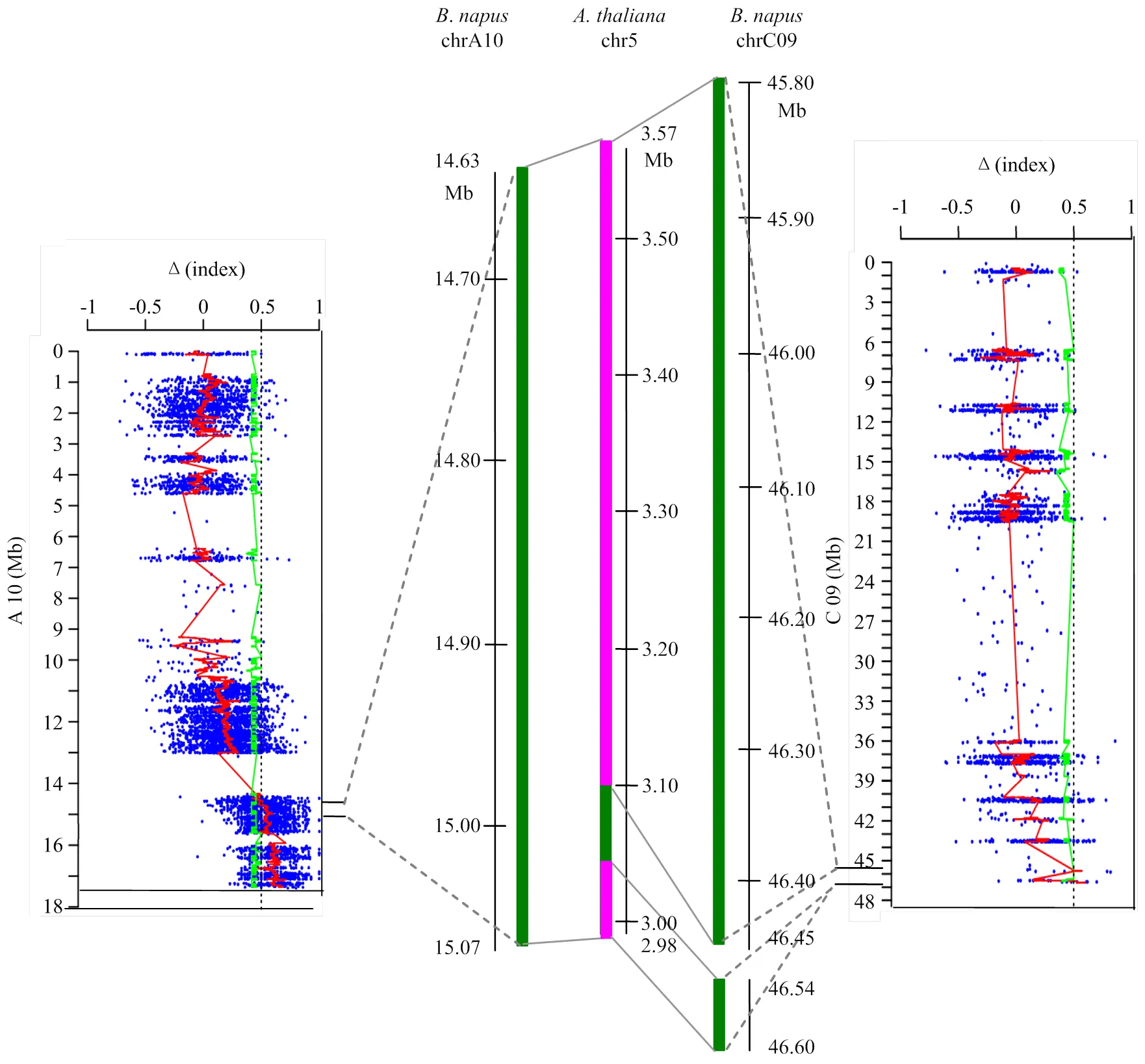
左右两边绿色线条代表A10关联区间部分区段和C09关联区间所所对应的物理位置,中间红色和绿色线条为两者对应到拟南芥基因组上的物理位置,其中红色部分代表A10关联区间部分区段和C09关联区间对应到拟南芥基因组相同位置

表3 关联区间内的候选基因预测

箭头和虚线区域代表氨基酸序列发生变异的位置
3.2 di1突变体有限花序候选基因的预测
利用本研究定位到的7个关联区间总共预测得到8个潜在候选基因,包括1个、3个、2个、1个和1个。、、、和在拟南芥花序和花形态建成的过程中具有非常重要的作用[17,34-41](图5)。基因克隆揭示有限花序突变体中的、和发生了基因编码区的碱基序列变异,并最终导致氨基酸序列变异。根据前人研究报道,属于磷脂酰乙醇胺结合蛋白家族,是维持花序分生组织特性所必需的,有抑制开花的作用[17],因此,在植物从营养生长到生殖生长这一成花转化过程中扮演非常重要的角色[42-44]。在拟南芥中,的突变将导致花序分生组织转化为花分生组织,形成单个的顶生花,最终使得花序由无限花序变为有限花序[11,17,45]。在豌豆和豇豆中的作用均被揭示为维持花序分生组织的无限生长习性,该基因的突变将产生有限花序类型的豌豆和豇豆[13,21]。此外,金鱼草[12]、番茄[18]、烟草[19]、黑麦草[20]、柑橘[22]、水稻[23]、苹果[24]等多个物种中均报道了由同源基因变异导致的有限花序突变。本研究中,位于A10染色体主效关联区域内,并且该基因在野生型和突变体中存在编码区和蛋白序列的变异,因此,推断对控制油菜有限花序变异具有关键作用。是自主开花途径中的一个关键基因,编码了一个同源基因,跟组蛋白的脱乙酞化复合体的形成有关。它的存在对FLC染色体组蛋白去乙酞化是必要的,FLC染色体组蛋白去乙酞化后,FLC由活化状态转变为非活化状态,从而启动植物开花[46]。刘新庆等[47]将拟南芥突变体与野生型相比较发现在野生型植株正常开花的阶段突变体植株尚未发育出花序,因此认为突变体影响了拟南芥花序的发育,并最终导致晚花表型。位于光周期途径,编码AP2/EREBP转录因子,但其作用却独立于,是通过与miR172的结合来抑制表达,最终抑制成花[48]。尽管这些基因位于不同的途径,但均可以作用于下游的花序分生组织特异基因,最终参与到花序和花的形态建成(图5)。
4 结论
油菜有限花序性状受2对隐性基因和1对隐性上位抑制基因互作控制。有7个区间与有限花序性状显著关联。和被推断为有限花序性状的候选基因。
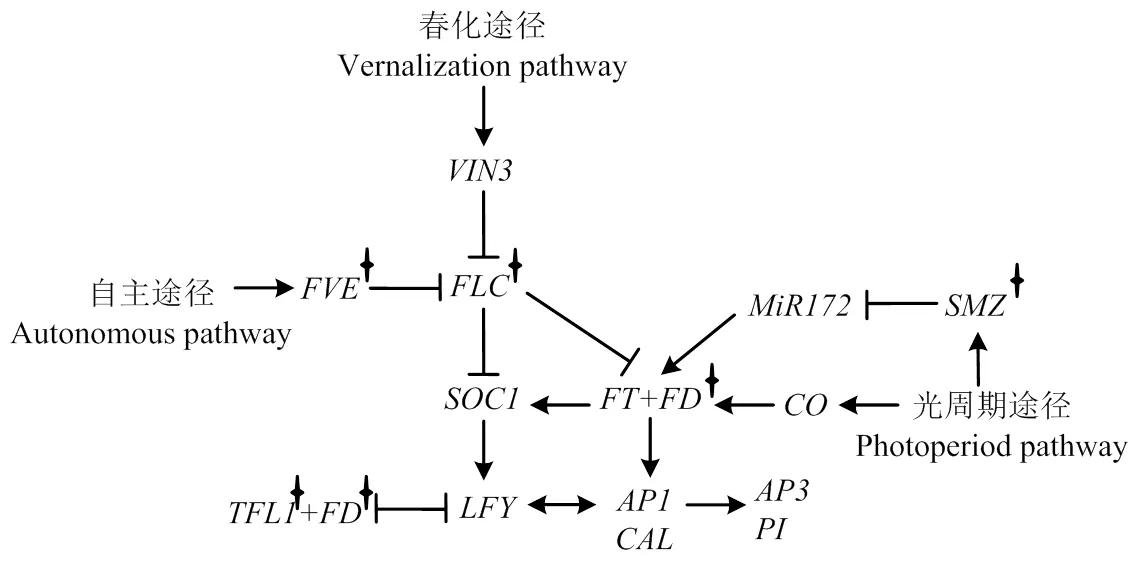
箭头表示促进作用,线段表示抑制作用;基因右上角星号代表候选基因
[1] 王汉中, 殷艳. 我国油料产业形势分析与发展对策建议. 中国油料作物学报, 2014, 36(3): 414-421.
WANG H Z, YIN Y. Analysis and strategy for oil crop industry in China., 2014, 36(3): 414-421. (in Chinese)
[2] 张凤英. 被子植物花序的演化问题. 植物杂志, 1985, 3: 040.
ZHANG F Y. The evolution of the inflorescence of angiosperms., 1985, 3: 040 (in Chinese)
[3] BANGA S S. Genetic manipulations for oilseeds improvement- conventional., 2007, 17: 34.
[4] HUIJSER P, SCHMID M. The control of developmental phase transitions in plants., 2011, 138(19): 4117-4129.
[5] LONG J, BARTON M K. Initiation of axillary and floral meristems in., 2000, 218(2): 341-353.
[6] HEPWORTH S R, KLENZ J E, HAUGHN G W. UFO in theinflorescence apex is required for floral-meristem identity and bract suppression., 2006, 223(4): 769-778.
[7] KWIATKOWSKA D. Flowering and apical meristem growth dynamics., 2008, 59(2): 187-201.
[8] HEMPEL F D, FELDMAN L J. Specification of chimeric flowering shoots in wild-type., 1995, 8(5): 725-731.
[9] AMASINO R. Seasonal and developmental timing of flowering., 2010, 61(6): 1001-1013.
[10] BRATZEL F, TURCK F. Molecular memories in the regulation of seasonal flowering: from competence to cessation., 2015, 16(1): 192.
[11] SHANNON S, MEEKS-WAGNER D R. A mutation in thegene affects inflorescence meristem development., 1991, 3(9): 877-892.
[12] BRADLEY D, CARPENTER R, COPSEY L, VINCENT C, ROTHSTEIN S, COEN E. Control of inflorescence architecture in., 1996, 379(6568): 791-797.
[13] DHANASEKAR P, REDDY K S. A novel mutation in TFL1 homolog affecting determinacy in cowpea ()., 2015, 290(1): 55-65.
[14] KAUR H, BANGA S S. Discovery and mapping ofgene associated with determinate plant growth habit., 2015, 128(2): 235-245.
[15] ZHANG H, MIAO H, LI C, WEI L, DUAN Y, MA Q, KONG J, XU F, CHANG S. Ultra-dense SNP genetic map construction and identification ofgene controlling the determinate growth habit inL.., 2016, 6: 31556.
[16] 刘毅. 有限开花习性芝麻的发现及其杂交后代. 中国油料, 1990, 1: 88.
LIU Y. The discovery of a sesame with determinate flowering habit and its hybrid progenies., 1990, 1: 88 (in Chinese)
[17] BRADLEY D, RATCLIFFE O, VINCENT C, CARPENTER R, COEN E. Inflorescence commitment and architecture in., 1997, 275(5296): 80-83.
[18] PNUELI L, GUTFINGER T, HAREVEN D, BEN-NAIM O, RON N, ADIR N, LIFSCHITZ E. Tomato SP-interacting proteins define a conserved signaling system that regulates shoot architecture and flowering., 2001, 13(12): 2687-2702.
[19] AMAYA I, RATCLIFFE O J, BRADLEY D J. Expression of CENTRORADIALIS (CEN) and CEN-like genes in tobacco reveals a conserved mechanism controlling phase change in diverse species., 1999, 11(8): 1405-1417.
[20] JENSEN C S, SALCHERT K, NIELSEN K K. A TERMINAL FLOWER1-like gene from perennial ryegrass involved in floral transition and axillary meristem identity., 2001, 125(3): 1517-1528.
[21] FOUCHER F, MORIN J, COURTIADE J, CADIOUX S, ELLIS N, BANFIELD M J, RAMEAU C. DETERMINATE and LATE FLOWERING are two TERMINAL FLOWER1/CENTRORADIALIS homologs that control two distinct phases of flowering initiation and development in pea., 2003, 15(11): 2742-2754.
[22] PILLITTERI L J, LOVATT C J, WALLING L L. Isolation and characterization of a TERMINAL FLOWER homolog and its correlation with juvenility in citrus., 2004, 135(3): 1540-1551.
[23] ZHANG S, HU W, WANG L, LIN C, CONG B, SUN C, LUO D. TFL1/CEN-like genes control intercalary meristem activity and phase transition in rice., 2005, 168(6): 1393-1408.
[24] KOTODA N, WADA M. MdTFL1, a TFL1-like gene of apple, retards the transition from the vegetative to reproductive phase in transgenic., 2005, 168(1): 95-104.
[25] Bansal P, Kaur P, Banga S K, Banga S S. Augmenting genetic diversity inthrough its resynthesis using purposely selected diploid progenitors., 2009, 3: 41-45.
[26] Kaur H, Gupta S, Kumar N, Akhatar J, Banga S S. Progression of molecular and phenotypic diversification in resynthesized(L.) gene pool with determinate inflorescence., 2014, 199(3): 325-338.
[27] LI K, YAO Y, XIAO L, ZHAO Z, GUO S, FU Z, DU D. Fine mapping of thegene associated with determinate growth habit., 2018, 131(1): 193-208.
[28] LI H, DURBIN R. Fast and accurate short read alignment with Burrows-Wheeler transform., 2009, 25(14): 1754-1760.
[29] LI H, HANDSAKER B, WYSOKER A, FENNELL T, RUAN J, HOMER N, ABECASIS G M, DUEBIN R. The sequence alignment/ map format and SAMtools., 2009, 25(16): 2078-2079.
[30] MCKENNA A, HANNA M, BANKS E, SIVACHENKO A, CIBULSKIS K, KERNYTSKY A, GARIMELLA K, ALTSHULER D, GABRIE S, DALY M, DEPRISTO M A. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data., 2010, 20(9): 1297-1303.
[31] WANG K, LI M, HAKONARSON H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data., 2010, 38(16): e164.
[32] TAKAGI H, ABE A, YOSHIDA K, KOSUGI S, NATSUME S, MITSUOKA C, UEMURA A, UTSUSHI H, TAMIRU M, TAKUNO S, INNAN H, CANO L M, KAMOUN S, TERAUCHI R. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations., 2013, 74(1): 174-183.
[33] CONESA A, GÖTZ S, GARCÍA-GÓMEZ J M, TEROL J, TALÓN M, ROBLES M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research., 2005, 21(18): 3674-3676.
[34] SEARLE I, HE Y, TURCK F, VINCENT C, FORNARAL F, KROBER S, AMASINO R A, COUPLAND G. The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in., 2006, 20(7): 898-912.
[35] BLÁZQUEZ M A, AHN J H, WEIGEL D. A thermosensory pathway controlling flowering time in., 2003, 33(2): 168-171.
[36] TAOKA K, OHKI I, TSUJI H, FURUITA K, HAYASHI K, YANASE T, YAMAGUCHI M, NAKASHIMA C, PURWESTRI Y A, TAMAKI S. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen., 2011, 476(7360): 332-335.
[37] NAN H Y, CAO D, ZHANG D Y, LI Y, LU S J, TANG L L, YUAN X H, LIU B H, KONG F J. GmFT2a and GmFT5a redundantly and differentially regulate flowering through interaction with and upregulation of the bZIP transcription factor GmFDL19 in soybean., 2014, 9(5): e97669.
[38] PURWESTRI Y A, OGAKI Y, TAMAKI S, TSUJI H, SHIMAMOTO K. The 14-3-3 protein GF14c acts as a negative regulator of flowering in rice by interacting with the florigen Hd3a., 2009, 50(3): 429-438.
[39] WIGGE P A, KIM M C, JAEGER K E, BUSCH W, SCHMID M, LOHMANN J U, WEIGEL D. Integration of spatial and temporal information during floral induction in., 2005, 309(5737): 1056-1059.
[40] JAEGER K E, PULLEN N, LAMZIN S, MORRIS R J, WIGGE P A. Interlocking feedback loops govern the dynamic behavior of the floral transition in., 2013, 25(3): 820-833.
[41] JUNG J H, SEO Y H, SEO P J, REYES J L, YUN J, CHUA N H, PARK C M. The GIGANTEA-regulated microRNA172 mediates photoperiodic flowering independent of CONSTANS in., 2007, 19(9): 2736-2748.
[42] HO W W H, WEIGEL D. Structural features determining flower promoting activity of., 2014, 26(2): 552-564.
[43] LI Q, FAN C M, ZHANG X M, WANG X, WU F Q, HU R B, FU Y F. Identification of a soybean MOTHER OF FT AND TFL1 homolog involved in regulation of seed germination., 2014, 9(6): e99462.
[44] HANANO S, GOTO K.TERMINAL FLOWER1 is involved in the regulation of flowering time and inflorescence development through transcriptional repression., 2011, 23(9): 3172-3184.
[45] TAHERY Y, ABHUL-HAMID H, TAHERY E. Terminal Flower 1 (TFL1) homolog genes in dicot plants., 2011, 12(545): 551.
[46] LIU F, QUESADA V, CREVILLE P, BAURLE I, SWIEZEWSKI S, DEAN C. TheRNA-Binding protein FCA requires a lysine-specific demethylase 1 homolog to downregulate FLC., 2007, 28: 398-407.
[47] 刘新庆, 郭蕊, 范晓宁, 王雪, 龙鸿. 拟南芥晚花突变体和营养生长时相转变. 热带农业科学, 2016, 36(6): 45-49.
LIU X Q, GUO R, FAN X N, WANG X, LONG H. Vegetative phase change in late-flowering mutantsand., 2016, 36(6): 45-49 (in Chinese)
[48] MATHIEU J, YANT L J, MURDTER F, KUTTNER F, SCHMID M. Repression of flowering by the miR172 target SMZ.gy, 2009, 7(7): e1000148.
(责任编辑 李莉)
Location and Mapping of the Determinate Growth Habit ofby Bulked Segregant Analysis (BSA) Using Whole Genome Re-sequencing
ZHANG YaoFeng, ZHANG DongQing, YU HuaSheng, LIN BaoGang, HUA ShuiJin, DING HouDong, FU Ying
(Institute of Crop and Nuclear Technology Utilization, Zhejiang Academy of Agricultural Sciences, Hangzhou, 310021)
【Objective】Mechanical harvesting has been one of the major goals of rapeseed breeding and genetic research worldwide. A natural and novel rapeseed mutant with determinate inflorescence()was identified in this study. Genetic analysis, gene mapping, candidate gene prediction and gene cloning were used to elucidate the genetic control of determinate inflorescence.【Method】For genetic analysis, reciprocalcrosses were performed between determinate inflorescence line FM8 and wild type FM7, and the inflorescence morphology was observed for F1and F2progenies. Two pools with 202lines and 20 wild type lines were constructed. For gene mapping of determinate inflorescence, 20× and 10× depth of whole genome re-sequencing were conducted for the two pools and parental lines, respectively. The associated loci were aligned to the genome offor synteny blocks searching. Potential candidate genes for determinate inflorescence were predicted by annotation analyses of genes within the physical boundaries of the associated regions.Gene cloning was used to identify polymorphisms and screen candidate gene(s). 【Result】Themutant showed a single fruiting body or a cluster of fruiting bodies at the top of the inflorescence axis, and the growth of inflorescence was hampered. The F1progenies from the reciprocalcrosses were indeterminate, and the trait segregation of indeterminate inflorescence and determinate inflorescence among F2progenies fit the 13:3 segregation ratio, assuming that the determinate inflorescence was controlled by two pairs of recessive duplicate genes interacting with one pair of recessive epistatic inhibitor genes. Whole genome re-sequencing of two pools and two parental lines identified 30123 homozygous SNPs and 107636 homozygous InDels. Seven significantly associated loci were mapped on chromosomes of A08, A09, A10, C08 and C09. Of which, the locus on chromosome A10 not only exhibited the highest peak, but alsoshowed homologous with the two loci on chromosome C09 by synteny analysis. Genes of(),(),(),() and() were predicted as potential candidate genes.Gene cloning identified coding region polymorphisms and protein polymorphisms for genes of,and.【Conclusion】The determinate inflorescence ofmutant was controlled by two pairs of recessive duplicate genes interacting with one pair of recessive epistatic inhibitor genes. Seven loci were significantly associated with determinate inflorescence. Of which, the loci on chromosomes A10 and C09 were homologous.,andshowed coding region polymorphisms and protein polymorphisms, and were deduced to be candidate genes for determinate inflorescence.
; determinate inflorescence; gene mapping; associated regions; candidate genes
2018-01-18;
2018-05-28
浙江省农业(粮食)新品种选育重大科技专项-油料新品种选育(2016C02050-8)、国家重点研发计划“七大农作物育种”(2016YFD0101306)、2017年农业科技发展项目(10102000317CC1201G/005)
张尧锋,E-mail:y.f.zhang@163.com。
傅鹰,Tel:0571-86404096;E-mail:fy97@163.com
10.3864/j.issn.0578-1752.2018.16.001
猜你喜欢
亚热带农业研究(2022年1期)2022-08-08
植物研究(2022年4期)2022-08-04
今日农业(2021年21期)2021-11-26
今日农业(2021年14期)2021-10-14
今日农业(2021年7期)2021-07-28
农业科技通讯(2021年1期)2021-03-06
中国农业科技导报(2020年3期)2020-03-15
中国食品学报(2019年12期)2019-01-13
果农之友(2018年5期)2018-10-19
科学种养(2017年6期)2017-06-13
