
组蛋白去乙酰化酶抑制剂阻断ERK/MAPK信号通路抗急性T淋巴细胞白血病的机制研究
2019-12-02 01:22沈莉菁刘嘉颖
医学研究杂志 2019年10期
高 晓 沈莉菁 刘嘉颖 李 香
急性T淋巴细胞白血病(T-cell acute lymphoblastic leukemia,T-ALL)好发于青少年,以常伴纵膈巨块,胸腔积液,易浸润中枢神经系统、肝脏、脾脏等组织,进展凶险,常规化疗不敏感著称。其发病机制涉及多种基因突变、信号通路转导和表观遗传因子改变[1]。近年来,随着化疗、免疫治疗及造血干细胞移植等综合治疗手段的发展,T-ALL患者的整体生存率有所改善,但仍有相当部分患者为复发难治[2,3]。因此,迫切需要寻找更有效的治疗靶点。
近年来研究发现,肿瘤细胞组蛋白乙酰化水平高的T-ALL患者,提示预后较好[4]。但多数患者存在组蛋白去乙酰化酶(histone deacetylase,HDAC)的异常高表达,导致组蛋白乙酰化水平较低,因此,笔者推测HDAC可能是T-ALL的潜在治疗靶点[5]。本研究旨在通过公共数据库挖掘与分析T-ALL中HDAC各亚型的表达情况,针对异常高表达HDAC亚型,选用选择性HDAC抑制剂西达本胺进行体外实验,探讨西达本胺对T-ALL细胞株的作用及可能的分子机制,为HDAC抑制剂在T-ALL的临床应用,提供理论依据。
材料与方法
1.材料:人急性T淋巴细胞白血病细胞株A3和Molt-4(上海中国科学院细胞库)。西达本胺由深圳微芯生物科技股份有限公司惠赠;二甲基亚砜(DMSO)购自美国MP Biomedicals公司;RPMI 1640培养基、胎牛血清、青霉素和链霉素混合液购自美国Gibco公司;CCK-8试剂盒为日本同仁化学研究所产品;Annexin V-FITC/PI细胞凋亡检测试剂盒为凯基生物公司产品;蛋白酶抑制剂、细胞裂解液NP-40、Pierce ECL蛋白发光底物试剂盒为美国Thermo Scientific公司产品,兔抗GAPDH、乙酰化H3K9(acetylated H3K9,Ac H3K9)、Mcl-1、BAX、多聚腺苷二磷酸核糖聚合酶(poly ADP-ribose polymerase,PARP)、剪切型PARP(cleaved PARP)、细胞外信号调节激酶1/2 (extracellular signal-regulated kinase1/2,ERK1/2)、磷酸化ERK1/2(phosphorylated ERK1/2, p-ERK1/2)单克隆抗体,鼠抗Bcl-2、半胱氨酸天冬氨酸蛋白酶9 (cysteinylasparate specific proteinase 9, caspase-9)单克隆抗体,山羊抗兔、山羊抗鼠HRP标记二抗均为美国Cell Signaling Technology公司产品;西达本胺溶于DMSO配制成10mmol/L储存液,-20℃保存,使用前用培养基稀释至工作浓度,使DMSO终浓度<1%。
2.方法:(1)数据库挖掘和分析:Oncomine数据库(https://www.oncomine.org)中分别检索HDAC1-11亚型在急性白血病与正常对照组中的表达情况,并进行统计学分析。(2)细胞培养:A3和Molt-4细胞均使用同一批次含10%胎牛血清、1% 100U/L青霉素、1% 100mg/L链霉素的RPMI 1640培养液,置于37℃、5%CO2且饱和湿度的细胞培养箱中,每隔1~2天更换培养液1次,倒置相差显微镜下观察,维持密度在(2~10)×105/ml。取对数生长期细胞用于实验。(3)CCK-8检测A3和Molt-4细胞增殖活力:取对数生长期的A3和Molt-4细胞株,按1×104个/孔细胞接种于96孔板中,设置空白组、对照组及实验组(西达本胺以100μmol/L为起始浓度,3倍梯度稀释,使终浓度分别为0.045、0.130、0.410、1.200、3.700、11.000、33.000、100.000μmol/L),每孔总体积为100μl,每组设5个复孔,置于37℃、5%CO2饱和湿度培养箱内分别孵育24、48、72h后,每孔避光加入10μl CCK-8溶液,将培养板放回培养箱中继续孵育2.5h后,用酶标仪测定在450nm处的吸光度(A)值。细胞存活率(%)=[(A实验组-A空白组)/(A对照组-A空白组)]×100%。应用Graphpad Prism 7.0软件计算药物的IC50。(4)流式细胞术检测A3和Molt-4细胞凋亡:取对数生长期的A3和Molt-4细胞株于6孔板,每孔接种细胞3×105个,实验组加入西达本胺使其终浓度分别为0.5、1.0、2.0μmol/L,同时设置对照组,37℃、5% CO2条件下培养48h。离心收集细胞,PBS洗涤2次,各组加入150μl Binding buffer重悬细胞后,实验组依次加入Annexin V-FITC和PI各5μl(Annexin V-FITC单染管只加入5μl Annexin V-FITC,PI单染管只加入5μl PI)混匀后室温避光孵育15min,最后各组再加入350μl Binding buffer轻轻混匀,1h内上机检测。(5)免疫蛋白印迹(Western blot)法检测组蛋白H3K9乙酰化、凋亡及ERK/MAPK信号通路蛋白水平:实验组西达本胺(终浓度为0.5、1.0、2.0μmol/L)分别处理A3和Molt-4 48h,同时设置对照组,PBS洗涤2次后收集细胞,加入含1%蛋白酶抑制剂cocktail的NP-40细胞裂解液提取总蛋白,冰上裂解30min后超声3次,5秒/次,收集总蛋白上清,用BCA法进行蛋白定量,加样至4%~10%的梯度SDS-PAGE凝胶中,经电泳、湿转至NC膜、5% BSA封闭、一抗4℃孵育过夜、TBST洗膜3次、二抗孵育、TBST洗膜3次后,ECL显影成像。使用Image J软件对各条带进行灰度分析,并以目的蛋白与内参蛋白灰度值的比值作为目的蛋白的相对表达水平。
结 果
1.HDAC1~11各亚型在T-ALL患者骨髓标本中的表达情况:本研究通过对Oncomine数据库(https://www.oncomine.org)进行深入挖掘,分析比较急性髓性白血病、急性B淋巴细胞白血病、T-ALL患者相对于正常对照组中HDAC1~11各亚型的表达情况。Ⅰ类HDAC1、2、3和Ⅱ类HDAC9、10在T-ALL组表达水平较正常对照组显著升高(HDAC1:t=8.398,P=2.38×10-7;HDAC2:t=6.976,P=2.41×10-6;HDAC3:t=3.359,P=0.002;HDAC9:t=6.814,P=5.55×10-6;HDAC10:t=9.493,P=6.72×10-8);HDAC5、6、8、11表达水平在T-ALL中与正常对照组比较,差异无统计学意义。
2.西达本胺对T-ALL细胞株增殖的影响:本研究使用选择性HDAC抑制剂西达本胺(靶点为HDAC1、2、3、10)和T-ALL细胞株A3、Molt-4进行后续实验。西达本胺(终浓度为0.045、0.130、0.410、1.200、3.700、11.000、33.000、100.000μmol/L)分别处理A3和Molt-4细胞株24、48、72h后,两种细胞存活率均随药物浓度和处理时间的增加而明显降低(P<0.05,图1),呈时间-浓度依赖性。西达本胺作用A3和Molt-4细胞株24h后的IC50值分别为2.24±0.18μmol/L和1.68±0.32μmol/L,48h的IC50值分别为0.66±0.10μmol/L和0.61±0.04μmol/L,72h的IC50值分别为0.29±0.13μmol/L和0.43±0.20μmol/L。

图1 CCK-8检测不同浓度西达本胺处理后A3和Molt-4细胞增殖曲线A.A3;B.Molt-4;与0μmol/L比较,*P<0.05,**P<0.01 (n=3)
3.西达本胺对T-ALL细胞株组蛋白H3K9的乙酰化水平的影响:本研究使用0.5、1.0、2.0μmol/L西达本胺分别处理A3和Molt-4细胞48h,组蛋白H3K9的乙酰化水平均较基线明显升高,呈浓度依赖(P<0.05,图2)。

图2 Western blot法检测不同浓度西达本胺对A3和Molt-4细胞组蛋白H3K9乙酰化水平的影响Ac H3.乙酰化的组蛋白H3K9;H3.内参蛋白;与0μmol/L比较,*P<0.01(n=3)
4.西达本胺对T-ALL细胞株凋亡的影响:0.5、1.0、2.0μmol/L西达本胺分别作用于细胞株A3和Molt-4 48h后,Annexin V-FITC/PI双染法检测细胞凋亡率。西达本胺诱导A3细胞凋亡的比率随浓度增加而递增(图3A),分别为14.60%±0.89%、17.11%±1.96%、26.51%±1.18%,与对照组比较,差异有统计学意义(P<0.05)。Molt-4细胞获得了类似结果(图3B),分别为4.74%±1.20%、5.76%±1.36%、10.48%±0.91%,与对照组比较,差异有统计学意义(P<0.05)。

图3 流式细胞术检测不同浓度西达本胺对A3和Molt-4细胞凋亡的影响A.A3;B.Molt-4;与0μmol/L比较,*P<0.05,**P<0.01(n=3)
5.西达本胺诱导T-ALL细胞株凋亡的可能机制:0.5、1.0、2.0μmol/L西达本胺分别作用于细胞株A3和Molt-4 48h后,Western blot法检测结果显示,与对照组比较,抗凋亡蛋白Bcl-2和Mcl-1均显著下调(P均<0.05),促凋亡蛋白BAX、线粒体凋亡途径蛋白活化型caspase-9、剪切型PRAP均显著上调(P均<0.05,图4)。Western blot法检测与细胞凋亡相关的ERK/MAPK信号通路蛋白,与对照组比较,p-ERK1/2蛋白水平均明显降低(P均<0.05),且0.5μmol/L的西达本胺即可明显下调A3细胞株的p-ERK1/2蛋白水平(P<0.05,图5)。

图4 Western bolt法检测不同浓度西达本胺对A3和Molt-4细胞相关凋亡蛋白表达的影响A.A3;B.Molt-4;Cleaved caspase-9.半胱氨酸天冬氨酸蛋白酶9;Cleaved PARP.剪切型多聚腺苷二磷酸核糖聚合酶;以各凋亡蛋白与内参蛋白GAPDH灰度值的比值作为蛋白相对表达水平;A:A3;B.Molt-4;与0μmol/L比较,*P<0.01(n=3)
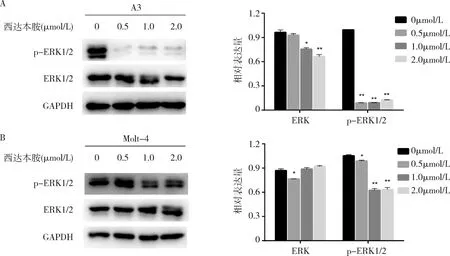
图5 Western bolt法检测不同浓度西达本胺对A3和Molt-4细胞中ERK1/2通路蛋白表达的影响A.A3;B.Molt-4;ERK1/2 细胞外信号调节激酶1/2;p-ERK1/2 磷酸化ERK1/2;以各通路蛋白与内参蛋白GAPDH灰度值的比值作为蛋白相对表达水平;与0μmol/L比较,*P<0.05,**P<0.01(n=3)
讨 论
组蛋白乙酰化在染色体重塑和调节转录活性方面发挥重要作用,主要受HDAC和组蛋白乙酰化转移酶的共同调节。其中,HDAC可降低组蛋白的乙酰化水平,使染色质结构变得致密从而抑制基因转录,促进肿瘤的发生。
HDAC异常高表达与T-ALL的发生有着密切的关系。文献报道,急性白血病患者中组蛋白低乙酰化并与不良预后相关[5];Gruhn等研究发现,与正常骨髓和外周血比较,ALL患者中Ⅰ类HDAC1、2、8高表达,这解释了之前研究中组蛋白低乙酰化的结果;此外,Moreno等发现HDAC1和2高表达与T细胞表型相关;Cardoso等[6]发现T-ALL患者可经染色体易位使转录因子TAL1过表达,进而招募含HDAC1的复合物促进T-ALL的发生。以上研究结果均提示Ⅰ类HDAC在T细胞白血病的发生中扮演着重要角色,也为T-ALL的治疗提供了新思路。本研究通过数据库挖掘分析发现Ⅰ类HDAC1、2、3和Ⅱ类HDAC9、10在T-ALL中显著高表达,结果与文献报道基本一致。
HDAC抑制剂具有广泛的抗肿瘤能力和较低的毒副作用,因此,临床上常作为多种血液肿瘤及实体瘤常规放、化疗手段的补充[7~9]。我国自主研发的新型HDAC抑制剂西达本胺主要针对ClassⅠ类的HDAC1、2、3以及Class Ⅱ类的HDAC10,目前的临床适应症为复发难治性外周T细胞淋巴瘤。其作用机制和疗效,与肿瘤类型密切相关[10,11],总体上可通过增加组蛋白H3和H4,以及非组蛋白p21、p53、C-MYC等乙酰化水平,调控相应基因转录,诱导细胞周期阻滞、促进肿瘤的分化、诱导细胞凋亡等发挥抗肿瘤作用,但在T-ALL中具体作用及机制研究较少。
本研究中西达本胺不仅对细胞株A3和Molt-4的增殖有明显的抑制作用,而且在不同时间点的IC50接近,抑制作用均呈浓度、时间依赖性的方式。随着西达本胺浓度的增加,T-ALL细胞株中组蛋白H3K9乙酰化水平明显增加,提示在表观遗传方面,西达本胺可有效抑制HDACs,增加组蛋白乙酰化水平。除抑制白血病细胞株增殖外,本研究选取西达本胺48h IC50左右的浓度进行细胞凋亡率的检测,发现西达本胺可显著诱导A3和Molt-4细胞的凋亡,且随浓度而递增。
在凋亡信号传递过程中,Bcl-2家族中抗凋亡蛋白和促凋亡蛋白共同影响线粒体膜的通透性,调控细胞色素C等细胞因子释放,激活caspase-9,再激活下游caspase效应分子,切割多种蛋白分子,如DNA损伤修复蛋白PARP,执行细胞凋亡的生物功能[12]。本研究发现,西达本胺可明显下调细胞株A3和Molt-4中抗凋亡蛋白Bcl-2和Mcl-1,上调促凋亡蛋白BAX、活化型caspase-9和剪切型PARP蛋白表达,这表明西达本胺可经线粒体途径介导T-ALL细胞凋亡。
肿瘤细胞的生长受到多种细胞外信号调控,调控细胞增殖及凋亡的经典信号通路促分裂原活化蛋白激酶 (mitogen-activated protein kinase,MAPK),受特定蛋白(ERK1/2、JNK、p38)的磷酸化活化形式介导,是多种肿瘤发病和转移的分子基础。在T-ALL中,ERK1/2/MAPK信号通路的异常激活是主要发病机制之一,而JNK/MAPK或p38/MAPK信号通路在T-ALL中无明显异常改变[13]。T-ALL患者很少发生ERK突变,但大约35%的成人T-ALL患者存在ERK1/2的持续激活,且与高白细胞数、对类固醇类药物反应差等不良预后因素有关[14]。ERK1/2的过度活化可影响肿瘤细胞的凋亡信号传递,主要通过间接影响细胞增殖通路NF-κB以及直接调控凋亡蛋白和caspase依赖的凋亡途径抑制细胞凋亡的发生[15,16]。因此,ERK1/2信号通路受抑可促进细胞凋亡,发挥抗肿瘤作用[17]。本研究发现,西达本胺可明显下调细胞株A3和Molt-4中p-ERK1/2蛋白的表达,因此,笔者推测西达本胺诱导T-ALL细胞凋亡的作用,可能与抑制ERK1/2信号通路的活化有关。
综上所述,针对T-ALL中高表达的Ⅰ类和Ⅱ类HDACs,本研究通过体外细胞实验表明西达本胺能有效抑制T-ALL细胞株A3和Molt-4细胞的增殖,增加组蛋白H3K9乙酰化水平,并通过调控凋亡蛋白表达和激活线粒体途径诱导细胞凋亡,其抗肿瘤机制可能与抑制ERK/MAPK信号通路转导有关,这为选择性HDAC抑制剂用于治疗T-ALL提供了部分理论和实验依据。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
中国药理学通报(2022年2期)2022-02-24
实用药物与临床(2020年8期)2020-12-27
中成药(2018年3期)2018-05-07
中华皮肤科杂志(2016年2期)2016-11-06
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
上海故事(2015年4期)2015-08-26
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年16期)2015-03-01
中国当代医药(2015年16期)2015-03-01
