
蛋白激酶A在常见慢性病病理过程中调控作用的研究进展*
2021-02-05 01:02徐海燕
中国病理生理杂志 2021年1期
徐海燕, 林 翠, 秦 虹
(中南大学湘雅公共卫生学院营养与食品卫生学教研室,湖南长沙410078)
蛋白激酶A(protein kinase A,PKA)是广泛存在于真核细胞中的一种丝氨酸/苏氨酸蛋白激酶,能够控制细胞增殖、分化及基因表达等多种细胞活动,与临床上多种疾病的发生发展密切联系[1]。随着经济水平的提高、生活环境的变化及社会人口老龄化,各种慢性病的发病风险日益严重。由于慢性病的病因复杂,具有发病率高、致残率高、致死率高及病情迁延难愈等特点,严重降低了人们的生命质量并给社会带来沉重的经济负担[2],因此对慢性病的防治显得尤为重要。最近许多研究显示,PKA 信号网络参与肥胖症、2 型糖尿病(type 2 diabetes mellitus,T2DM)、恶性肿瘤、骨质疏松症(osteoporosis,OP)及阿尔茨海默病(Alzheimer disease,AD)等常见慢性病的发病机制,影响这些疾病的发生和发展,因此,明确PKA在这些疾病中的作用对于寻找疾病的潜在治疗靶点具有重要意义。本文就近年来PKA调控常见慢性病发病过程的研究进展进行综述。
1 PKA的分子结构特征
哺乳动物大约有2%的基因组编码蛋白激酶,这些蛋白激酶通过调节基因转录、细胞分裂及分化等多种细胞活动,参与生物体的生长发育过程,是治疗多种疾病的主要靶点[3]。蛋白激酶家族中最简单的成员之一是PKA,有I型(PKA-I)和II型(PKA-II)两种同工酶。PKA 全酶是由两个催化(C)亚基和两个调节(R)亚基组成的一个异四聚体(图1)。PKA-I和PKAII的C亚基相同,但R亚基分别为RI和RII,而且R亚基又有两种亚型(Rα和Rβ),C亚基有四种亚型(Cα、Cβ、Cγ和PRKX)[4]。PKA全酶是没有催化活性的,其活化依赖第二信使环磷酸腺苷(cyclic adenosine monophosphate,cAMP)的刺激。R 亚基上包含两个cAMP结合位点,两个cAMP分子结合到R亚基上使R亚基的构象发生变化,释放出C 亚基,从而使得PKA具有催化活性,见图1[5]。此外,PKA的功能特异性依赖于其亚细胞定位,PKA通过R亚基与细胞内不同位点的A 激酶锚定蛋白(A-kinase anchoring proteins,AKAPs)靶向结合并接近特异性底物,有活性的PKA C 亚基通过磷酸化这些下游底物来调节真核细胞中的生理功能[3]。目前已证实有250多种PKA的磷酸化底物,所有的这些蛋白组成了哺乳动物细胞中的PKA信号网络[6]。PKA 在机体内错综复杂的信号网络参与了多种慢性病发病机制的调控,因此PKA可能成为治疗这些疾病的关键信号分子。
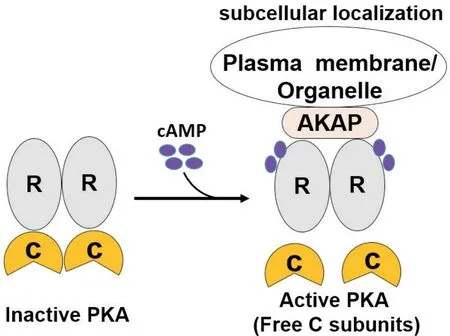
Figure 1.Molecular structure and activity regulation of PKA.图1 PKA的分子结构和活性调节
2 PKA在5种常见慢性病中的作用
2.1 PKA 对肥胖症发病的抑制作用 肥胖症是由于长期能量摄入超过能量消耗,从而引起脂肪组织中大量脂肪堆积所致,与冠心病、高血压及非酒精性脂肪肝等许多慢性疾病的发病有关,对心理健康也有负面影响[4]。目前,肥胖症的主要治疗方式是通过抑制食欲,限制肠内的营养吸收,促进脂肪组织的脂质代谢及能量消耗等方面进行干预。
研究证实,PKA 信号的激活能够抑制食欲以减少能量摄入。机体的摄食中枢下丘脑通过分泌神经肽Y(neuropeptide Y,NPY),从而刺激摄食行为,降低机体的产热效应[7]。有研究显示,在下丘脑中注射cAMP 激动剂或PKA 激动剂后,下丘脑中cAMP/PKA 信号通过磷酸化激活cAMP 反应元件结合蛋白(cAMP-response element binding protein,CREB)从而抑制NPY基因表达,可减少 NPY 诱导的摄食行为[8]。瘦素是一种脂肪来源的激素,可通过与下丘脑瘦素受体结合,激活PKA/CREB/NPY 通路,从而减少食欲并有效预防肥胖症[9]。
活化的PKA 可通过抑制脂质合成,促进脂质分解、氧化及产热等多方面改善肥胖症。在脂质合成方面,活化的PKA 通过磷酸化激活肝X 受体α(liver X receptor α,LXRα),减少LXRα 与脂质合成的关键转录因子固醇调节元件结合蛋白1c(sterol regulatory element binding protein-1c,SREBP-1c)的启动子结合,导致 SREBP-1c 蛋白表达量减少[10]。此外,活化的PKA 也可以通过直接磷酸化SREBP-1c 的Ser308位点,促进SREBP-1c 的泛素化降解,从而抑制脂质合成相关酶的转录[11]。在脂质分解方面,活化的PKA 能够磷酸化脂滴上的脂滴包被蛋白5(perilipin 5)和比较基因识别58(comparative gene identification-58,CGI-58),促使 CGI-58 从 perilipin 5 上解离,导致脂肪甘油三酯脂酶(adipose triglyceride lipase,ATGL)活化,从而启动脂解的第1 步[4]。而且,活化的PKA 可磷酸化激素敏感性脂酶(hormone-sensitive lipase,HSL)的Ser563、Ser659 及Ser660 位点,p-HSL进入脂滴,进一步催化脂解反应[12]。在脂质氧化方面,活化的PKA 可以通过提高sirtuin 1(SIRT1)的活性而激活过氧化物酶体增殖物激活受体γ 辅激活因子 1α(peroxisome proliferator-activated receptor γ coactivator-1α,PGC1α)及其下游的转录因子过氧化物酶体增殖物激活受体α(perixisome proliferator-activated receptor α,PPARα),从而促进脂肪酸氧化相关基因的转录[13]。活化的 PKA 还能够磷酸化 AMP 活化蛋白激酶(AMP-activated protein kinase,AMPK)从而增加其下游脂肪酸β-氧化关键酶肉毒碱棕榈酰基转移酶1α(carnitine palmityl transferase-1α,CPT1α)的表达,促进脂肪酸氧化[14]。在产热方面,PKA 是介导 β3-肾上腺素能受体(β3-adrenergic receptor,β3-AR)信号诱导白色脂肪褐变及产热的关键分子,脂肪细胞膜上的β3-AR 激活后继而通过PKA/CREB传导途径,促进PGC1α 和解偶联蛋白1(uncoupling protein 1,UCP1)等与产热有关的基因的转录,导致脂肪细胞产热增加,脂肪组织中脂质蓄积减少[15]。上述研究揭示了活化的PKA 可通过抑制食欲、促进脂质代谢及产热耗能等而减轻肥胖症。在当前的高热量饮食环境下,肥胖率越来越高,但目前缺乏治疗肥胖症的有效手段,靶向激活PKA 可能成为治疗肥胖症的一个新的研究方向。
2.2 PKA 对T2DM 的组织差异性调控作用 T2DM是一种慢性进行性疾病,表现为胰岛素分泌不足和胰岛素抵抗引起的血糖水平持续升高,如果血糖长期得不到控制将引起微血管、大血管及神经的病变,进而危害心脏、肾脏、眼睛等重要器官[16]。目前,T2DM 无法治愈,其主要的缓解方法就是进行血糖控制,而机体内血糖水平由胰岛、肝脏、骨骼肌、脂肪和大脑等多种组织共同调节[17]。
活化的PKA 信号能够促进胰岛β 细胞分泌胰岛素来降低血糖水平。在胰岛β 细胞中,活化的PKA能够磷酸化ATP敏感钾通道、葡萄糖转运蛋白2(glucose transporter 2,GLUT2)、L 型电压门控钙通道和胰岛素分泌性囊泡相关蛋白来促进胰岛素转录和分泌[17]。肌腱膜纤维肉瘤癌基因同源物A(musculoaponeurotic fibrosarcoma oncogene homolog A,MAFA)是一种刺激胰岛素表达的转录因子,已有研究证实,激活PKA/CREB 信号通路后可促进MAFA 的表达从而增加胰岛素的分泌[18]。
肝脏中PKA信号通路也在血糖调控中起到重要作用。在正常生理状态下,肝脏可将餐后血糖转化为糖原储存,空腹时,胰高血糖素和肾上腺素与G 蛋白偶联受体结合而激活肝细胞中cAMP/PKA 信号通路而减少糖原合成,并分解肝糖原释放葡萄糖到血液中,从而维持血糖的平稳;在T2DM中,肝胰岛素抵抗及胰高血糖素信号传导异常会导致肝葡萄糖输出升高[19]。研究显示,抗T2DM药二甲双胍可通过抑制小鼠肝细胞中cAMP/PKA信号通路传导降低血糖水平。PKA活性降低后,能够减少糖原磷酸化酶激酶(glycogen phosphorylase kinase,GPK)和磷酸果糖激酶2(phosphofructokinase-2,PFK2)的磷酸化从而减少糖原分解和糖异生[19-20]。此外,PKA 活性降低后还可减少下游CREB的磷酸化水平,在转录水平上降低磷酸烯醇丙酮酸羧化激酶(phosphoenolpyruvate carboxykinase,PEPCK)和葡萄糖-6-磷酸酶(glucose-6-phosphatase,G6P)的基因表达来抑制肝糖异生,从而减少肝脏葡萄糖输出,有效发挥抗T2DM作用[21]。
PKA 信号也可通过促进骨骼肌糖代谢下调血糖水平。骨骼肌也能将血糖转化为糖原储存,但骨骼肌中由肌糖原分解产生的葡萄糖并不是释放到血液中,而是作为骨骼肌运动的主要能量来源。骨骼肌运动时,肾上腺素激活肌细胞中β2-AR/cAMP/PKA信号级联反应,活化的PKA 继而促进GLUT4 转移到质膜来增加肌细胞对葡萄糖的摄取[22],并且进一步激活SIRT1/PGC1α 通路促进脂肪酸氧化和能量消耗而改善骨骼肌胰岛素抵抗,从而降低血糖水平[23]。此外,骨骼肌中PKA的活化,也能抑制糖原合酶活性从而减少肌糖原合成,并激活糖原磷酸化酶促进糖原分解供能[24]。
脂肪组织中PKA信号也参与了血糖的调控。肥胖状态下脂肪组织中活化的PKA 通过磷酸化激活HSL 催化脂肪分解并释放过多的游离脂肪酸(free fatty acid,FFA),而血清FFA水平过高能够诱发全身性胰岛素抵抗导致血糖升高[25]。二甲双胍能够抑制脂肪细胞中PKA 信号来抑制脂解作用,从而提高胰岛素敏感性来下调血糖水平[26]。
大脑中的PKA信号通路能够与外周组织协调控制葡萄糖稳态。在下丘脑神经元中,胰高血糖素、瘦素和胰岛素等激素能通过激活cAMP/PKA 通路抑制肝脏葡萄糖产生而减少血糖水平[17,27]。胰高血糖素激活PKA 后,活化的PKA 可直接磷酸化KATP通道的Kir6.2/SUR1 亚单位而激活 KATP通道[28],而瘦素和胰岛素则是在激活PKA 后,通过AMPK/丙二酰-CoA/CPT-1/LCFA-CoA/PKC-δ 脂质感应途径引起 KATP通道的活化,然后通过刺激迷走神经的肝分支,激活胰岛素受体底物(insulin receptor substrate,IRS)/磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)信号传导途径而提高肝胰岛素敏感性,继而减少肝糖异生因子PEPCK 和G6P 的表达,抑制肝脏葡萄糖产生[27,29]。上述研究显示 PKA 信号对 T2DM 的调控作用具有组织差异性,因此,如何针对性地综合调控人机体内PKA 的活性使其发挥最大的有益作用,仍是一个待研究的问题。
2.3 PKA 对不同恶性肿瘤的差异性调控作用 恶性肿瘤由于其细胞处于低分化状态,细胞分裂及生长迅速,不受正常生理调节,破坏正常组织与器官。恶性肿瘤的治疗手段主要是通过阻碍细胞分裂增殖并诱导细胞凋亡来抑制细胞的无限生长。
PKA 信号能够从多方面抑制有些恶性肿瘤细胞增殖。cAMP 类似物dbcAMP 通过激活多形胶质母细胞瘤细胞中的PKA,活化的PKA 进一步激活下游CREB/PGC1α 途径来促进线粒体生物合成和细胞分化,从而抑制胶质瘤细胞的分裂增殖[30]。PKA 也能干扰细胞的有丝分裂周期而抑制细胞增殖。在TT和MZ-CRC-1 两种甲状腺髓样癌细胞系中,cAMP 类似物8-Cl-cAMP 通过激活PKA,阻断了细胞有丝分裂的G2/M 期向G0/G1期转变和G0/G1期向S期的转变,从而抑制了体外甲状腺髓样癌细胞增殖。目前,8-Cl-cAMP 是有效的抗肿瘤药物之一,并作为抗癌药进入I/II期临床试验阶段[31]。而且,PKA 能够作用于细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)信号通路来调节细胞增殖。ERK 是丝裂原激活蛋白激酶(mitogen-activated protein kinase,MAPK)家族的一员,调节着细胞的增殖、分化和存活,在许多恶性肿瘤中都可以发现ERK 的过度激活[32]。在 MDA-MB-231 乳腺癌细胞和 HEK293 细胞中,活化的PKA 能够通过直接磷酸化Raf-1,也可以通过磷酸化Src 来激活Rap1,从而阻止Raf-1 与Ras的结合,导致ERK 失活和细胞增殖减少[33-34]。而且,活化的PKA 通路也可以通过抑制ERK 活性而减少血管内皮生长因子表达,进而抑制结直肠癌组织中血管生成来阻止癌细胞生长[35]。
但PKA在一些其他细胞中却激活ERK而促进细胞增殖。在神经元海马细胞中,β2-AR/cAMP/PKA信号途径能够激活ERK 维持突触可塑性和记忆[36]。BRaf 是大脑中的主要的Raf 亚型,活化的PKA 可以通过Rap1/B-Raf途径激活ERK而促进GH3垂体瘤细胞增殖[37]。在T细胞中,活化的PKA通过磷酸化造血蛋白酪氨酸磷酸酶(hematopoietic protein tyrosine phosphatase,HePTP)使其失活,从而导致ERK 从HePTP/ERK 复合体中释放而激活ERK[38]。这些研究证实PKA对不同恶性肿瘤细胞增殖的调控存在差异。
PKA 不仅调控细胞增殖,也能诱导恶性肿瘤细胞凋亡。在MCF-7 人乳腺癌细胞中,活化的PKA 能激活Fas/FasL 凋亡信号通路并增加死亡受体4 表达直接导致细胞凋亡,还能磷酸化激活转录因子4(activating transcription factor 4,ATF4)和p38 MAPK,引起内质网应激而诱导细胞凋亡,而且活化的PKA 还可以磷酸化抑癌蛋白p53 的Ser15 位点而促进p-p53的核易位,导致细胞周期停滞和细胞凋亡[39]。
恶性肿瘤细胞的无限增殖和恶性转化与PKA-I/PKA-II比率增加有关。PKA-II的表达主要存在于正常的非增生组织中,而恶性肿瘤细胞中能检测到高水平的PKA-I,而且原癌基因Ras 的突变能够诱导PKA-I 及其调节亚基 RIα 的表达增加[40-41],PKA-I 的过表达被认为是大多数人类恶性肿瘤的特征性标志,因此,PKA-I 可能作为一种新的生物标志物用于恶性肿瘤检测。
2.4 PKA 对OP 发病的抑制作用 OP 是老年人中常见的一种疾病,随着人口老龄化加剧,OP 发病率越来越高。OP 是骨形成与骨吸收间不平衡,引起骨质减少、骨骼脆性增加和骨组织微结构破坏为特点的一种全身代谢性骨病,其引起的慢性疼痛、骨骼畸形和骨折会严重影响老年人的生活质量,甚至导致死亡[42]。成骨细胞和破骨细胞维持骨代谢平衡,成骨细胞增殖减弱和破骨细胞增殖增强是OP 的重要发病机制,因此,促进成骨分化并抑制破骨分化是治疗OP的关键要素[43]。
体内外研究已证明PKA信号通路可促进成骨细胞的分化而有效改善OP。在小鼠骨折模型及成骨样MC3T3-E1细胞中,激活PKA通路使得细胞内的p-CREB 表达增加,一方面,通过提高碱性磷酸酶(alkaline phosphatase,ALP)活性以增加细胞基质钙化,并通过激活骨形成蛋白(bone morphogenetic protein,BMP)/Smad 信号通路来促进转录因子Runx2 表达来诱导成骨细胞特异性基因的转录,从而促进细胞的成骨分化;另一方面,p-CREB 进入细胞核能够促进骨桥蛋白和骨钙素等成骨相关因子表达来促进成骨作用[44-45]。而且,在去卵巢的 OP 小鼠模型中,活化的PKA信号能够磷酸化激活信号传导及转录激活蛋白3(signal transducer and activator of transcription 3,STAT3),然后刺激骨髓巨噬细胞极化为M2亚型,M2型巨噬细胞在骨表面分泌转化生长因子β1(transforming growth factor-β1,TGF-β1),从而促进骨髓基质细胞(bone marrow stromal cells,BMSCs)向骨表面迁移[46],而 BMSCs 中活化的 PKA 进一步磷酸化激活成骨分化的的关键信号分子β-catenin 的Ser675位点并促进β-catenin 核易位,随后募集LEF/TCF DNA 结合因子来启动成骨细胞分化相关基因的表达,从而促使BMSCs 分化为成骨细胞并抑制BMSCs 的成脂分化,从而改善小鼠的OP 症状[47]。另外,BMSCs 中活化的PKA 也能通过直接磷酸化β-catenin的Ser552和Ser675 两个位点或激活PI3K/Akt信号以增加磷酸化的糖原合成酶激酶3β(glycogen synthetase kinase-3β,GSK3β)表达,从而抑制 β-catenin 的泛素化降解[48],维持BMSCs中β-catenin含量而促进成骨分化。
除了影响成骨细胞分化,PKA也能通过抑制破骨细胞分化来改善OP。研究显示,cAMP 类似物8-ClcAMP激活小鼠原代骨髓细胞中PKA/ERK1/2途径后,抑制了核因子κB(nuclear factor-κB,NF-κB)入核而抑制破骨细胞分化相关基因的转录表达[49]。活化的PKA 还能通过直接磷酸化破骨细胞祖细胞中的关键转录因子活化T细胞核因子胞浆1型(nuclear factor of activated T-cells,cytoplasmic 1,NFATc1)而阻止其核易位并抑制破骨细胞的分化,而且活化的PKA还通过增加p-GSK3β 和β-catenin 表达,诱导骨保护素(osteoprotegerin,Opg)的表达,进而抑制骨髓中破骨细胞前体的分化,减少骨吸收而增加骨含量[50-51]。目前的这些证据显示激活PKA信号通路可抑制OP的发展。
2.5 PKA 对AD 发病的抑制作用 AD 是常见的一种老年痴呆症,其典型的组织病理学特征包括淀粉样斑块的形成、突触损失和神经纤维缠结,从而导致进行性记忆丧失和认知功能障碍。目前,AD缺乏特效治疗方法,其症状的缓解可通过抑制大脑中淀粉样斑块积聚及改善突触可塑性。
PKA 信号通路能够减少神经元中淀粉样斑块形成而改善AD。AD 动物模型和体外培养的神经元中PKA/SIRT1 信号通路的激活,可上调去整合素金属蛋白酶10(a disintegrin and metalloprotease domain 10,ADAM10)并下调β-位点淀粉样前体蛋白裂解酶1(β-site amyloid precursor protein cleaving enzyme 1,BACE1)两种酶的表达以抑制淀粉样前体蛋白衍生为Aβ,抑制神经元中Aβ 积累,从而减轻AD 症状[52]。然而也有研究表明,PKA 信号通路会加速AD 的病理过程。胰岛素降解酶(insulin-degrading enzyme,IDE)不仅能够催化胰岛素降解,也能够促进Aβ的降解。在T2D 与AD 混合小鼠模型中观察到,活化的PKA 通过减少IDE 表达引起Aβ 积聚和神经元凋亡而加速AD 的病理进展[53],这种矛盾的情况可能是因为小鼠模型不同而引起。
PKA 信号通路也通过增强突触可塑性及减少神经元凋亡来改善AD。突触损失及功能障碍是AD 痴呆症状严重程度的主要的衡量指标。PKA 信号通路除了改善Aβ 诱导的突触损失外,研究显示,活化的PKA 可增强兴奋性突触传递。兴奋性突触传递是由AMPA 型谷氨酸受体(AMPA-type glutamate receptors,AMPARs)介导的,海马CA1 神经元处的cAMP/PKA 被激活后,活化的PKA 一方面通过磷酸化激活含GluA1 亚基的AMPAR 促进了该受体向突触运输,一方面通过磷酸化激活含GluA3 亚基的AMPAR 增加了突触间的电信号传递,激活了突触可塑性,从而促进大脑的学习、记忆以及记忆巩固[54-55]。脑源性神经 营 养 因 子(brain-derived neurotrophic factor,BDNF)是神经营养素家族的一员,可促进神经元的存活、分化和生长,调节突触活动,参与维持大脑海马区的学习和记忆功能,海马区中BDNF 的减少,是AD 认知功能下降的原因之一[56]。大鼠海马区PKA/CREB 信号通路的激活,可上调BDNF 表达并抑制神经元凋亡,从而改善海马区大脑的长期记忆及认知功能障碍,对AD 具有重要保护作用[57]。上述研究证实大脑中PKA的激活可能缓解AD的临床症状。
3 结语
PKA 经cAMP 激活后可磷酸化下游不同的底物蛋白而调控多条信号通路,由此发挥多种生理功能,与机体各种生命活动密切相关。疾病的发生发展往往受到细胞内错综复杂的信号通路调控。随着PKA在肥胖症、T2DM、恶性肿瘤、OP、AD等多种疾病中研究的不断展开,证实有选择性的激活PKA 信号通路对这些疾病可能都有一定的治疗作用。人体作为一个整体,PKA 信号通路在机体内的作用具有组织特异性,而且又能调控同一细胞内多条信号通路发挥多种功能,因此PKA 在疾病发病的分子机制中不完全是一个正向作用。目前cAMP 依赖的PKA 信号与上述相关疾病的确切关系仍不完全清楚,故需进一步研究深入探讨PKA 在这些疾病发生发展、防治过程中发挥作用的内在机制,这不仅有助于从整体上了解PKA 通路在疾病中的调控作用,还可为这些疾病的临床治疗提供新的治疗靶点。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
云南化工(2020年11期)2021-01-14
世界最新医学信息文摘(2020年68期)2020-12-25
作文成功之路·小学版(2020年6期)2020-07-27
天津医科大学学报(2019年6期)2019-08-13
延安大学学报(医学科学版)(2019年1期)2019-03-29
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
遗传(2014年3期)2014-02-28
中国医学科学院学报(2013年6期)2013-03-11
