
HDAC8 基因变异所致Cornelia de Lange 综合征1 例报告
2021-02-26 06:41史晓依徐瑞娟杜开先贾天明王丽君宋盼盼
临床儿科杂志 2021年1期
董 燕 史晓依 徐瑞娟 杜开先 贾天明 王丽君 李 肖 宋盼盼
郑州大学第三附属医院 1.神经内科,2.儿童发育行为科(河南郑州 450000)
Cornelia de Lange 综合征(Cornelia de Lange syndrome,CdLS)又称德朗热综合征、阿姆斯特丹侏儒症,是一种表现多样化的遗传性疾病,主要临床表现为特殊外貌、生长发育迟缓、多器官畸形、听力障碍等,呈常染色体显性或X 连锁显性遗传[1]。随着分子生物学技术的进步,最新文献报道发病率达1/30000~1/10000[2]。通过对万方、维普、知网及PubMed 等数据库进行搜索发现,与国外相比,国内文献报道仍较少,且大部分是临床诊断,郑州大学第三附属医院2019 年确诊1 例CdLS,基因检测提示为HDAC8基因新发变异,查阅ClinVar和HGMD数据库未收录此变异,未见相关文献报道,现报告如下。
1 临床资料
患儿,女,1 岁2 月龄,因发育落后11 个月就诊。患儿自3月龄起出现发育落后,少动、睡眠多。至就诊时患儿不会爬,不能独走,无意识发“mama”音,无自主觅食意识。患儿5个月竖头,8个月独坐。出生体质量2.5 kg,身长43 cm。父母均体健,无相似表现,否认近亲婚配,否认家族中有类似疾病发生。体格检查:体质量7.5 kg(<-2SD)头围41 cm(<-3SD),身长68 cm(<-3SD);神清,精神可,毛发浓厚,前囟已闭,脸小,弓形眉,眼距宽,眼裂小,鼻梁低平,嘴窄,唇红、薄,下颌短,喜笑;心肺及腹部无异常;外生殖器无异常(图1);手脚小,左手通贯掌,四肢肌力及肌张力无异常。入院查血常规、血生化、心脏彩超、头颅MRⅠ等未见异常。听力检查示声阻抗、耳声发射左侧未通过,脑干听觉诱发电位双侧阈值40 db,40 Hz听觉相关电位双侧阈值40 db。婴幼儿智能发育量表(Children’s Developmental Center of China,CDCC)测试示智能及运动发展指数均<50,智能及运动均相当于8.2月龄。
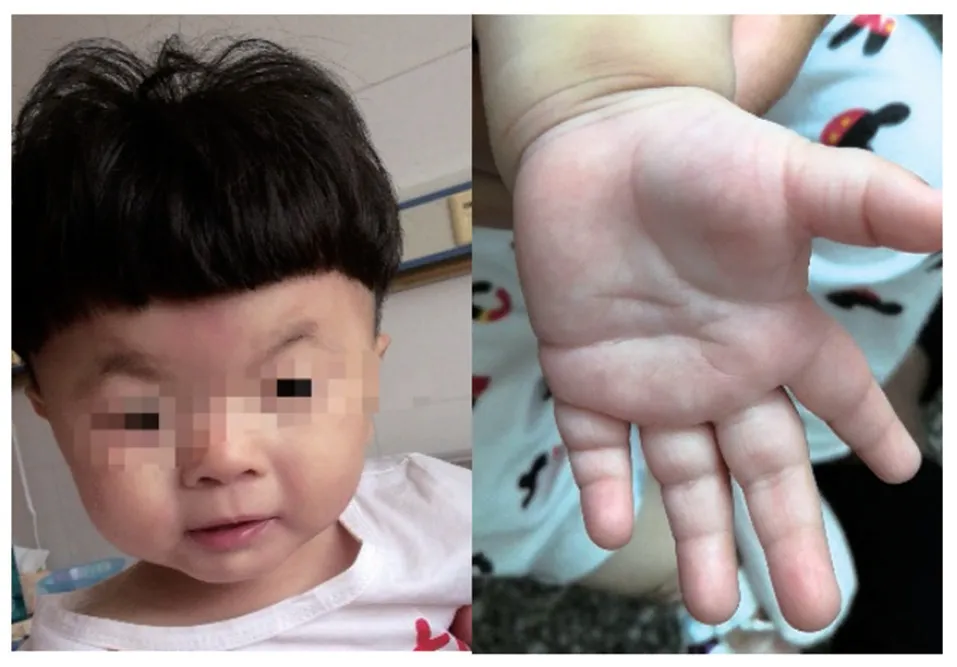
图1 患儿特殊面容及手纹异常表现
为明确病因,经医院医学伦理委员会批准,患儿父母知情同意,采集患儿及其父母外周血进行家系全外显子组测序及低深度全基因组测序。家系低深度全基因组测序未见异常。家系全外显子组测序发现,患儿HDAC8基因7号外显子c.675C>A(p.Y225X)存在无义变异,经过Sanger 验证发现父母无此变异,该位点为新发(de novel)变异(图2)。查阅ClinVar 和HGMD数据库此变异未收录,未见相关文献报道。根据美国医学遗传学与基因组学学会(ACMG)制定的遗传变异分类标准与指南进行致病性分析:①LOF变异导致基因功能可能丧失(超强致病证据,PVS1);②c.675C>A (家系)经双亲验证的新发变异,符合X连锁遗传病特征(强致病证据PS2);③MAF<0.005,dbSNP数据库、千人基因组数据库、EXAC数据库等未见收录,属于低频变异(中等致病证据,PM 2);④两种以上统计方法预测出变异对基因(基因产物)有影响,保守性及蛋白结构预测软件证据(MutationTaster,GERP,phyloP20way,phastCons20way)(可能致病证据,PP3)。综上,c.675C>A变异判定为致病性变异,证据强度为“PVS 1+PS 2+PM 2+PP 3”。先证者杂合de novo变异(父母均为野生型),符合X染色体显性遗传方式及家系共分离。根据临床资料、遗传模式及生物学致病等级评级确诊患儿为CdLS。
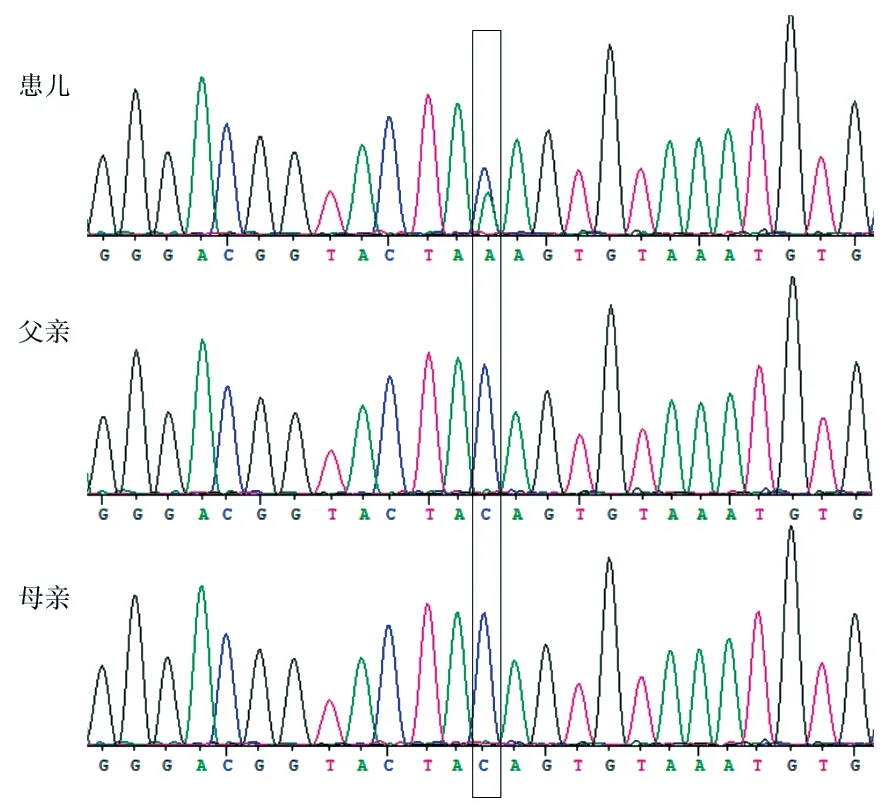
图2 患儿及父母HDAC8 基因Sanger 测序图
患儿治疗10天后出院,给予长期家庭训练。1岁8月龄会走,目前2岁5个月身高85 cm(-2SD),体质量9.5 kg(<-3SD);仅能发单双音节,理解力差,能听懂部分指令;步态无异常,能跑,精细抓物可。
2 讨论
CdLS 是一种多器官受累的遗传性疾病,1933 年由荷兰儿科医师Cornelia de Lange 首次报道。CdLS多为散发病例,少数存在家族发病倾向;男女患病比例约1:1.3[3]。研究显示,患儿诊断年龄为17 岁9 个月[4],仅7%于新生儿期确诊[5]。2019 年研究者对收录在PubMed、CⅠNAHL、Scopus等数据库中的病例进行meta分析,共纳入1 310例CdLS患儿,发现其主要临床表现是特殊面容和听力异常,少数有气道发育畸形所致睡眠呼吸障碍[6]。国内近几年报道较多,以“德朗热综合征”、“Cornelia de Lange综合征”、“HDAC 8mutation”为检索词在万方、维普、中国知网及PubMed进行检索,对报道的中国国内病例进行统计,共计46例患儿,连同本例患儿47 例。总结分析发现,国内CdLS的男女发病比例约1:1.6;足月出生27例、早产12 例,8 例未提及出生史;确诊年龄从新生儿期到13岁不等,其中18例(约38.3%)在新生儿期确诊。
CdLS 临床表现差异性大,部分患者出生时即存在特殊外貌,容易识别,但表现不典型者多因生长发育迟缓或智力低下就诊时诊断。依据临床表现的异质性CdLS 分为经典型、轻型和拟CdLS 型。CdLS 诊断主要依赖于典型的临床表现及基因检测。2018年国际CdLS专家小组联合制定了CdLS的诊断标准。临床诊断:至少3个主要特征,得分≥11提示经典CdLS,无论是否存在相关基因变异;如果存在至少2个主要特征,得分9~10 分表示非经典CdLS;如果至少存在1个主要特征,得分≥4 需注意,建议行分子检测;<4分则不足以怀疑CdLS。其中主要特征包括:连眉和/或浓眉;短鼻,鼻梁凹陷和/或鼻尖上翘;长和/或平的人中;上唇薄和/或口角下斜;少指和/或无指畸形;先天性膈疝。次要特征包括:整体发育迟缓和/或智力低下;宫内生长迟缓;宫外生长迟缓;小头畸形;手脚小;第5指短;多毛。每条主要特征得2 分,每条次要特征得1分[7]。本例患儿存在2条主要特征(鼻梁低平,唇薄),5 条次要特征(迟缓和/或智力低下,宫内生长迟缓,宫外生长迟缓,小头畸形,手脚小),得分9分,为非经典CdLS,因发育落后就诊,在诊疗过程中发现患儿特殊面容,临床表型较其他基因型轻。
根据基因变异不同,可将CdLS分为5型:NIPBL相关的常染色体显性遗传的1 型(OMⅠM:122470),SMC 1 A相关的X 连锁显性遗传的2 型(OMⅠM:614701),SMC 3相关的常染色体显性遗传的3 型(OMⅠM:610759),RAD21相关常染色体显性遗传的4 型(OMⅠM:300590)以及HDAC 8相关X 连锁显性遗传的5 型(OMⅠM:300882)[8]。以NIPBL基因变异多见,占60%~70%[7],HDAC8基因变异占4%左右[9]。
CdLS患者的临床表型与基因变异存在一定联系,黏连蛋白复合物参与姐妹染色单体聚合,在染色体分离、基因组稳定性等方面具有重要作用。SMC 1 A、SMC 3、RAD 21是构成黏连蛋白复合物的核心亚基,NIPBL、HDAC 8等是黏连蛋白复合物的调控组件。NIPBL基因变异产生截断效应时临床表型较重,而其错义变异以及携带HDAC8、NIPBL、SMC1A等框内缺失及错义变异的患者临床表型较轻[9]。
本文总结国内46 例病例,其中26 例患者行基因检测[1,2,8,10-21],NIPBL变异阳性率达76.9%,较文献报道比例更高。HDAC 8基因位于染色体Xq 13.1 区,编码Zn2+依赖的组蛋白去乙酰基转移酶[22],调节SMC3蛋白的去乙酰化,该基因变异可影响黏连蛋白复合物正常功能。自2012年首次报道HDAC8基因相关CdLS以来,国外仅报道70余例[7,22-23];国内学者的中英文文献目前仅检索到3例中国CdLS患者HDAC8基因变异报道[2,20-21]。与国外相比,国内CdLS研究报道仍较少,且大部分是临床诊断。本例患儿通过基因检测确诊,为HDAC8基因新发无义变异。
CdLS尚无特效治疗手段,主要在于防治并发症、重视产前诊断。CdLS 死因中以呼吸系统及消化系统疾病多见。
综上,CdLS的主要临床差异性大,部分患者存在特殊外貌,但部分不典型患者仅有生长发育迟缓或智力低下,相关致病基因的高通量测序或全外显子测序可协助诊断。本研究发现HDAC 8基因一个未见报道的变异位点。
猜你喜欢
临床肝胆病杂志(2020年1期)2020-12-20
支部建设(2020年15期)2020-07-08
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
中国科技纵横(2016年15期)2016-12-29
甘肃教育(2016年22期)2016-12-20
中学生理科应试(2016年4期)2016-11-19
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
百科知识(2015年18期)2015-09-10
中国石油大学学报(社会科学版)(2015年2期)2015-06-15
