
锰基氧化物活化过硫酸盐降解水中有机污染物的研究进展
2021-08-23 10:29王一凡李小蝶侯美茹王兆慧
环境科学研究 2021年8期
王一凡, 李小蝶, 侯美茹, 王兆慧,2,3*
1.华东师范大学生态与环境科学学院, 上海市城市化生态过程与生态恢复重点实验室, 上海 200241 2.上海有机固废生物转化工程技术研究中心, 上海 200241 3.自然资源部大都市区国土空间生态修复工程技术创新中心, 上海 200062
随着人类大规模的生产活动,生活污水和工业废水排放量的增加,水资源污染问题日益严重. 水体中的污染物主要包括重金属、难降解的有机物、植物营养物、放射性物质、石油类、病原体、悬浮物等,这些污染物由于自身特征与含水介质的作用,易从地表水源迁移进入地下水中造成严重污染,而且很难通过常规的废水处理方法有效去除[1].
锰基催化剂具有晶型结构及性质多变、比表面积大、毒性低、自然丰度高、环境友好[10]、成本低且可大规模制备等优势,作为水处理中SR-AOPs的催化材料,可用于活化PMS/PDS降解水中有机污染物. Mn(Ⅱ、Ⅲ、Ⅳ和Ⅶ)氧化物因其氧化态和结构具有多样性[11],对于过硫酸盐的活化效率亦存在较大差异. 因此,深入理解锰氧化物的类型结构和作用原理,对其用于活化PS降解污染物具有重要的理论和现实意义. 鉴于此,该文主要对锰基氧化物活化PS降解水中有机污染物的应用研究进行了综述. 概述了锰基氧化物的类型、结构特征、合成方法及影响反应活性的因素,针对各种锰基氧化物活化PS的反应机理进行了系统的阐述,最后提出建议和研究展望.
1 锰基氧化物的类型、结构特征及合成方法
1.1 类型与结构特征
锰氧化物在自然界中广泛存在,富含于海洋和陆地环境中. 作为天然存在的强氧化剂之一,广泛参与环境中各种化合物的氧化还原反应. 由于锰的价态十分丰富,因此其氧化物的存在形式也较为多样. 常见的锰氧化物有几十种,如软锰矿(MnO2)、褐锰矿(Mn2O3)、硬锰矿(mMnO·MnO2·nH2O)、水锰矿[MnO2·Mn(OH)2]等. 锰氧化物随氧化价态的升高,碱性降低,酸性增强. 同时,锰元素的氧化价态越高,其氧化物的催化活性越强. 锰氧化物通常具有较高的化学反应活性,可以通过吸附、氧化分解等形式对有机物的转化产生影响.
锰氧化物的晶体类型和形态结构是影响其催化反应性能的重要因素. 二氧化锰的晶体结构分为3类,即一维隧道结构、二维层状结构及三维网状结构,主要存在的晶体类型包括α-MnO2、β-MnO2、γ-MnO2、λ-MnO2、δ-MnO2等. 锰氧化物基本结构是由1个Mn原子和6个O原子配位形成的六方密堆积结构及立方密堆积结构,即[MnO6]八面体与邻近的八面体会共用棱与角顶,从而形成复杂的网格结构. 不同形态结构的锰氧化物,在化学反应过程中表现出不同的催化反应性能.
1.2 合成方法
常见合成锰基氧化物的方法有水热法[12]、溶剂热法[13]、溶胶凝胶法[14]、沉淀法[15]、微乳液法[16]等. 水热法是合成锰基材料的经典方法,可以从含有不同摩尔比的MnCl2和KMnO4的体系中制备α-MnO2、β-MnO2、γ-MnO2[17]. 3种晶体结构的MnO2在低温条件下均具有较高的催化反应活性. Xiao等[18]采用改进的水热法工艺,通过还原KMnO4,改变前驱物的浓度及水热的温度来制备3D分层结构的MnO2催化剂. Cao等[19]利用模板法和水热法制备4种不同形态(空心球、空心立方体、纳米棒、海胆)的Mn2O3,其中空心球和空心立方体结构的Mn2O3具有较强的稳定性及催化活性. Tian等[20]以高锰酸钾和氯化氢为原料,利用水热法在不同温度下分别合成不同形状(纳米管、纳米棒和纳米颗粒)的MnO2,其中纳米棒的MnO2具有较高的催化性能. Atiqueullah等[21]在含有KMnO4和甘油的体系中,利用溶胶-凝胶法制备Mn5O8(单斜晶和纳米棒结构)和Mn2O3的复合纳米颗粒. Meng等[22]在无任何催化剂或模板剂的情况下,利用低温水热法合成γ-MnOOH纳米棒,并以其为前驱物在高温煅烧下制备出β-MnO2纳米棒以及MnO、Mn2O3、Mn3O4纳米粒子.
2 影响反应活性的因素
锰基氧化物活化PDS或PMS受自身物理化学性质、外部反应条件的影响,包括反应温度、溶液pH、晶型结构、共存离子、污染物初始浓度、催化剂的用量等[23].
2.1 共存阴离子
水环境中通常存在大量的无机阴离子,如碳酸氢根(HCO3-)、硝酸根(NO3-)、亚硝酸根(NO2-)、氯离子(Cl-)、CO32-、硫酸根(SO42-)等. 在PMS氧化体系中这些无机阴离子可消耗或进一步产生活性自由基[24]. 例如,NO2-与SO4·-和·OH易发生反应〔见式(1)~(2)〕,与污染物竞争活性自由基,因此表现出比其他无机阴离子更强的抑制效果.
417 Progress in the detection of macular ganglion cell complex thickness
SO4·-+NO2-→NO2·+SO42-,k=8.8×108L/(mol·s)
(1)
·OH+NO2-→NO2·+OH-,k=8.0×109L/(mol·s)
(2)
共存离子SO42-和Cl-对MnO2/NF/PMS体系AO7降解动力学呈现不同的影响规律. SO42-存在的情况下,AO7的降解率未受明显影响,然而随着Cl-的添加量逐渐增多,染料降解呈现显著的加速趋势. HCO3-和CO32-能以不同速率与SO4·-和·OH反应,因此二者常作为自由基的主要清除剂,抑制有机污染物的降解[25]. 若环境中存在过量的天然有机质(NOM), 也会消耗SO4·-或·OH从而降低降解效率[26].
氯化物和碳酸盐对不同的活化过程表现出迥异的影响规律[27]. 在PMS/Mn3O4-MnO2体系共存阴离子会抑制污染物环丙沙星(CIP)的降解,抑制作用依次表现为CO32->HCO3->NO3->SO42-,而Cl-起促进降解的作用[28]. 原因可能在于HCO3-与CO32-将SO4·-和·OH 清除,并产生了氧化能力较弱的碳酸盐自由基[29]. Cl-直接与PMS发生反应生成活性氯(HClO/Cl2)或者Cl-与SO4·-和·OH反应生成氯自由基,对基于PMS氧化体系的影响程度受pH和Cl-浓度控制. Deng等[30]研究表明HCO3-和CO32-均抑制卡马西平(CBZ)的降解速率. 由于CO32-与SO4·-的反应速率约是HCO3-的4倍,因此CO32-相较于HCO3-对CBZ降解的抑制作用更明显. 此外,阴离子还可以通过竞争吸附作用影响污染物的降解效率. 一方面,通过占据催化剂表面的活性位点抑制催化剂与氧化剂的接触,影响自由基的生成效率;另一方面,较高浓度的阴离子会抑制污染物在锰氧化物表面的吸附.
2.2 溶液pH
溶液pH会改变锰基材料表面电荷分布,影响材料与PMS/PDS之间的相互作用及反应活性[31]. Jo等[32]研究氧化铁固定化MnO2复合材料活化PS时发现,pH为9.0时四氯化碳和苯的降解率最高. 但当pH较低时锰氧化物会溶解,Mn(Ⅱ)释放改变了Mn基材料的表面结构. 若存在一定量的质子与PMS/PDS 结合,也会干扰并抑制Mn基材料的催化反应活性[33]. 然而,Xiao等[34]发现pH为3.0~7.0时从含锰废水中合成纳米结构MnO2对苯酚的降解率较高. 原因在于:PMS在碱性条件下不稳定,易分解成O2和SO42-;酸性条件能够促进SO4·-和·OH的生成,且该条件下的自由基具备较高的氧化还原电位. 因此,随着初始pH的增加,污染物的去除率逐渐降低[35]. MnO2/UV/PMS体系降解4-氯苯酚也具有上述相同的反应趋势[36],即酸性条件下污染物的降解效率较高.
2.3 反应温度
反应温度是影响锰基氧化物活化PMS/PDS效率重要因素之一. Mn基氧化物活化PMS/PDS过程中,有机污染物的降解速率与温度呈正相关[37-38]. 原因在于升高温度,PMS/PDS自身的热活化、Mn基氧化物活化和自由基降解污染物的反应都会加速. 但反应温度并非越高越好,要根据催化剂类型、氧化剂稳定性和处理成本选择适宜的反应温度.
式(3)表示速率常数[k,L/(mol·s)]与温度之间的相关性,可用于计算活化能.
lnk=lnA-Ea/RT
(3)
式中:A为频率因子,L/(mol·s);Ea为活化能,J/mol;R为通用气体常数,J/(mol·K);T为绝对温度,K. 不同锰氧化物降解同一种污染物,会计算得出不同的活化能. 由于锰氧化物活化PMS/PDS 和SO4·-氧化降解苯酚等多个反应都受温度影响,因此计算得到的Ea其实为包含多个基元反应的表观活化能. 不同体系之间不宜采用Ea来比较锰氧化物的催化性能.
2.4 晶型结构
不同形态结构的锰基氧化物活化PMS时展现出不同的催化反应活性,例如,结晶态的α-MnO2具有较高的苯酚降解率[39]. 不同形状的Mn2O3晶体催化苯酚的降解速率依次表现为Mn2O3(正立方体)>Mn2O3(正八面体)>Mn2O3(八面体截断)[40]. Wang等[41]制备的一维纳米α-MnO2的催化活性表现为α-MnO2(纳米线)>α-MnO2(纳米棒)>α-MnO2(纳米管). 不同结晶相的一维MnO2纳米颗粒(α-MnO2纳米线、β-MnO2纳米棒、γ-MnO2纳米纤维)催化活性大小为α-MnO2>γ-MnO2>β-MnO2. α-MnO2的反应性和稳定性最高[42]. Huang等[43]建立了4种不同MnO2结构的催化反应活性与物理化学性质之间的线性关系,得出反应活性顺序依次为α-MnO2>γ-MnO2>β-MnO2>δ-MnO2. 此外,催化反应性还取决于Mn的氧化态,反应趋势表现为Mn2O3>MnO>Mn3O4>MnO2[44].
3 锰基氧化物活化PMS的机制
3.1 MnO2及其复合型氧化物活化PMS
MnO2活化PMS产生的SO4·-和·OH作为主要的氧化性物种能有效去除苯酚、BPA和有机合成染料等污染物〔见式(4)~(7)〕.
HSO5-+2MnO2→SO5·-+OH-+Mn2O3
(4)
HSO5-+Mn2O3→SO4·-+H++2MnO2
(5)
SO4·-+H2O→·OH+H++SO42-
(6)
SO4·-+OH-→·OH+SO42-
(7)

(8)
(9)
(10)
(11)
RhB+SO4·-→CO2+H2O+SO42-
(12)
除RhB外,BPA在PMS-MnO2体系氧化降解存在2种反应过程:一是MnO2的直接氧化[48-49],在酸性条件尤为明显;二是催化氧化,α-MnO2、β-MnO2以及γ-MnO2活化PMS均可生成SO4·-和·OH和1O2. Wang等[50]通过乙醇(EtOH)和叔丁醇(TBA)2种清除剂的自由基淬灭试验,证实SO4·-在活化降解过程中起主导作用. Dong等[51]揭示了β-MnO2纳米线具有良好的可分离性及显著的催化降解作用. Sui等[52]证明α-MnO2和β-MnO2单晶体纳米结构颗粒在去除亚甲基蓝的类芬顿反应中表现出优异的催化性能[53].
除上述单一锰氧化物外,α-MnO2负载Co3O4纳米颗粒可以有效非均相活化PMS降解水溶液苯酚〔见图1(a)〕[54]. 仅采用Co3O4对PMS进行活化需要使用专门制备的惰性载体,且反应活性过分依赖于载体. 而Co3O4与MnO2的复合对PMS的活化和苯酚的降解具有显著的协同作用. Co3O4/MnO2具有较高的氧化还原电位,能够加快SO4·-的生成,提高苯酚的降解速率. 因此,SO4·-的生成效率取决于Co3O4与MnO2的氧化还原反应.
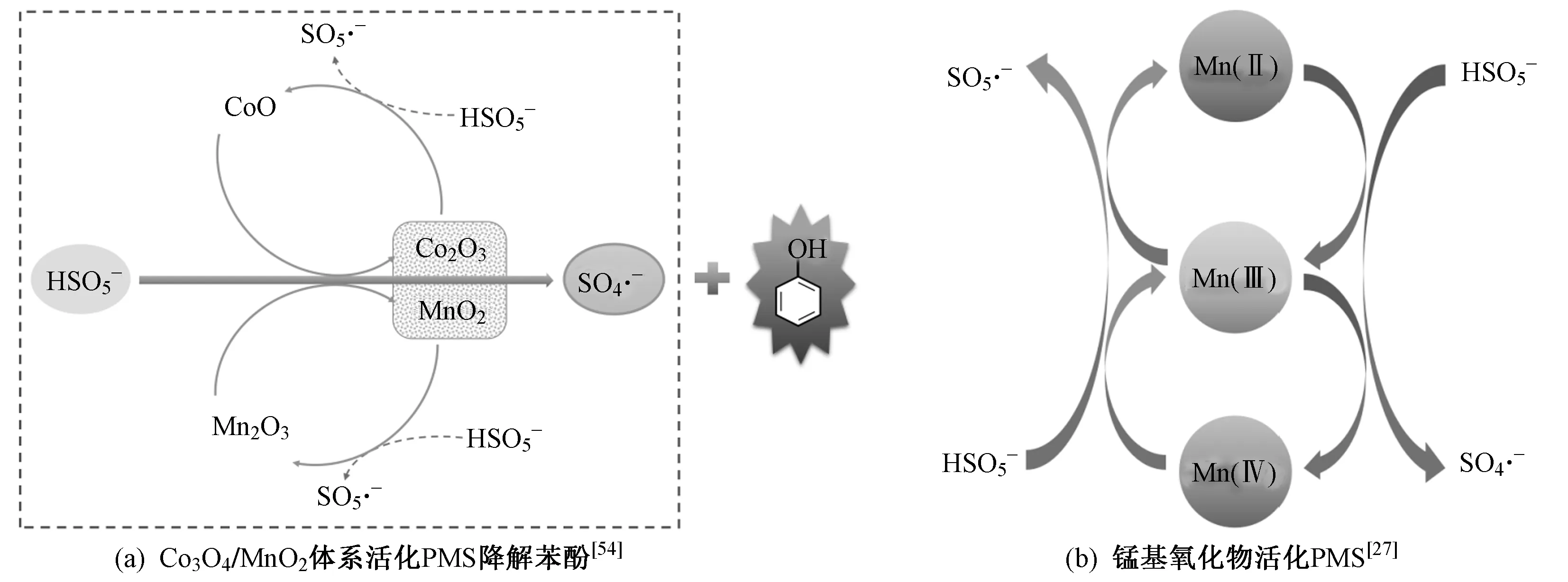
图1 锰基氧化物活化PMS的反应机理Fig.1 Reaction mechanism of PMS activation by manganese-based oxides
此外,淬灭试验和电子自旋共振(ESR)数据证明,除自由基机理外,非晶态的MnO2可通过非自由基机理活化PMS,生成PMS-MnO2络合物降解有机污染物[55]. 在非晶态MnO2和PMS之间形成的活性络合物可以介导BPA和PMS之间的直接电子转移,选择性地氧化酚类化合物. 因此,非晶态的MnO2/PMS 体系在有机化合物选择性氧化方面具有较好的应用前景.
3.2 MnOx及其复合型氧化物活化PMS
除MnO2外,其他锰基氧化物(MnO、Mn2O3、Mn3O4等)也可用于活化PMS,且相较于一元氧化物具有更好的催化效果以及高稳定性、低成本、多价态、高催化活性等优势,其反应机理〔见式(13)~(15)〕如下:
HSO5-+2MnO→SO4·-+H++Mn2O3
(13)
HSO5-+2Mn3O4→SO4·-+H++3Mn2O3
(14)
HSO5-+Mn2O3→SO4·-+H++2MnO2
(15)
近年的研究表明,具有混合Mn价态和不同纳米结构、形状等的锰氧化物复合材料,或与其他金属元素材料形成的新型复合材料可增强催化反应活性. Saputra等[56]合成的Mn3O4和Co3O4纳米颗粒活化降解效果显著,反应活性表现为Mn3O4>Co3O4>Fe3O4. 但金属离子Co2+具有毒性,对人体健康的危害较为严重,不推荐使用.
如上所述,锰氧化物催化剂在催化PMS的过程中除产生自由基外还可能会生成单态氧1O2[57-58]〔见式(16)(17)〕.1O2是O2的激发态,在光化学过程中通过光诱导使能量转移到O2而产生[59].1O2作为一种温和的选择性氧化剂,不易被常用的自由基捕获剂甲醇或叔丁醇等淬灭[60]. 除光诱导外,还可以通过PMS的自分解、羟基自由基的重组、表面吸附的O2获取能量[61]等方式产生1O2,如Zhou等[62-63]利用苯醌活化PMS生成1O2.
HSO5-+SO52-→HSO4-+SO42-+1O2
(16)
2·OH→1/21O2+H2O
(17)
Tian等[64]通过降解苯酚和四环素(TC)研究生物氧化锰(BioMnOx)和3D α-Mn2O3活化PMS的能力,试验证明BioMnOx/PMS体系同时产生了SO4·-和1O2.1O2是苯酚降解的主要活性氧物种,它的产生可能仅来自PMS和BioMnOx的相互作用. 因此,相较于3D α-Mn2O3,BioMnOx表现出较高的催化活性、降解效率和稳定性. Huang等[65]合成了新型Mn1.8Fe1.2O4纳米球,利用双金属氧化物中的2种不同金属(Mn和Fe)阳离子之间的协同作用催化PMS降解BPA,其催化性能明显高于Mn/Fe的单金属氧化物. Mn(Ⅱ)和Fe(Ⅱ)向PMS提供电子,Mn(Ⅲ)提供额外的单电子活化PMS. 随后,高价态的Mn和Fe被HSO5-还原,通过Mn和Fe氧化还原循环维持Mn1.8Fe1.2O4的催化作用〔见式(18)~(20)〕.
(18)
(19)
(20)
Sahoo等[66]曾提出Mn和Fe之间的电子转移是二者产生协同作用的主要原因. 大多数有机污染物的多相催化降解发生在氧化物的表面,因此反应底物对活性位点的预吸附(主要是络合效应)显得尤为重要[67-68]. 当磷酸盐与催化剂表面过渡金属发生配位而使其无法裸露于表面时,Fe/Mn协同作用被抑制,与Fe配位的磷酸盐取代了PMS,降低了降解效率. Yao等[69]证实磁性MnFe2O4纳米颗粒和MnFe2O4/还原氧化石墨烯(MnFe2O4-rGO)复合物在活化PMS降解水中有机染料(甲基紫、甲基橙、亚甲基蓝、橙Ⅱ和罗丹明B)时具有较高的活性和良好的稳定性. MnFe2O4纳米催化剂表面上的Fe(Ⅱ)/Mn(Ⅱ)与PMS反应生成SO4·-,Fe(Ⅲ)/Mn(Ⅲ)再被PMS还原生成更多Fe(Ⅱ)/Mn(Ⅱ)〔见式(21)~(24)〕. 类似于类Fenton反应[70-71],Mn(Ⅱ)/Mn(Ⅲ)和Fe(Ⅱ)/Fe(Ⅲ) 是生成SO4·-的氧化还原对.
(21)
(22)
(23)
(24)
除双金属氧化物之外,Zhang等[72]制备了具有磁性的复合新型三金属氧化物Co3MnFeO6纳米颗粒,作为非均相催化剂活化PMS降解有机污染物卡马西平(CBZ). 吸附到催化剂表面的HSO5-被还原性的Co(Ⅱ)、Mn(Ⅱ)、Mn(Ⅲ)、Fe(Ⅱ)活化,通过电子转移生成SO4·-. Co和Mn之间的氧化还原循环以及再生的Co2+和Mn2+会连续激活PMS,生成具有强氧化性的SO4·-,SO4·-与H2O或OH-之间相互作用生成·OH,与单金属氧化物或双金属氧化物相比,三金属氧化物的催化活性更高. Co、Mn和Fe在Co3MnFeO6催化剂中的协同作用是活化PMS高效生成SO4·-的主要原因. Co和Mn作为主要的活性位点,活化PMS产生自由基;Fe使Co3MnFeO6催化剂具有磁性,成为PMS和CBZ的吸附位点. 3种金属之间的氧化还原循环使Co3MnFeO6催化反应高效持续进行,表现出较高的催化性和稳定性[73]. 综上,锰基氧化物活化PMS的普遍机理与锰的氧化还原循环有关〔见图1(b)〕.
4 锰基氧化物活化PDS的机制
PMS水溶液呈酸性且活性自由基的产生量相对较低,而PDS水溶液通常呈中性,易储存且环境友好. 相较于活化PMS,除SO4·-和·OH在PDS体系中作为主要的反应物质外,还存在单独生成1O2的氧化反应路径.
4.1 MnO2活化PDS
与PMS反应机理相似,MnO2也被用于活化PDS降解硝基苯[74]和2,4-二氯苯酚[75]等污染物. SO4·-和·OH是α-MnO2活化PDS的主要活性物种. 然而,PDS/β-MnO2体系降解苯酚是一种非自由基的氧化反应[76]. 与β-MnO2活化PMS产生SO4·-、·OH和1O2不同,β-MnO2活化PDS仅产生1O2. Fang等[77-78]认为,1O2的产生可能是由于超氧离子和自由基的直接氧化或重组,MnO2的表面会形成一种中性的亚稳态锰中间体(MnⅣ—O—O—SO3),与S2O82-反应并伴随MnⅣ—O断裂形成超氧化物中间体O2·-[79],随后MnⅣ直接氧化O2·-生成1O2[80](见图2).
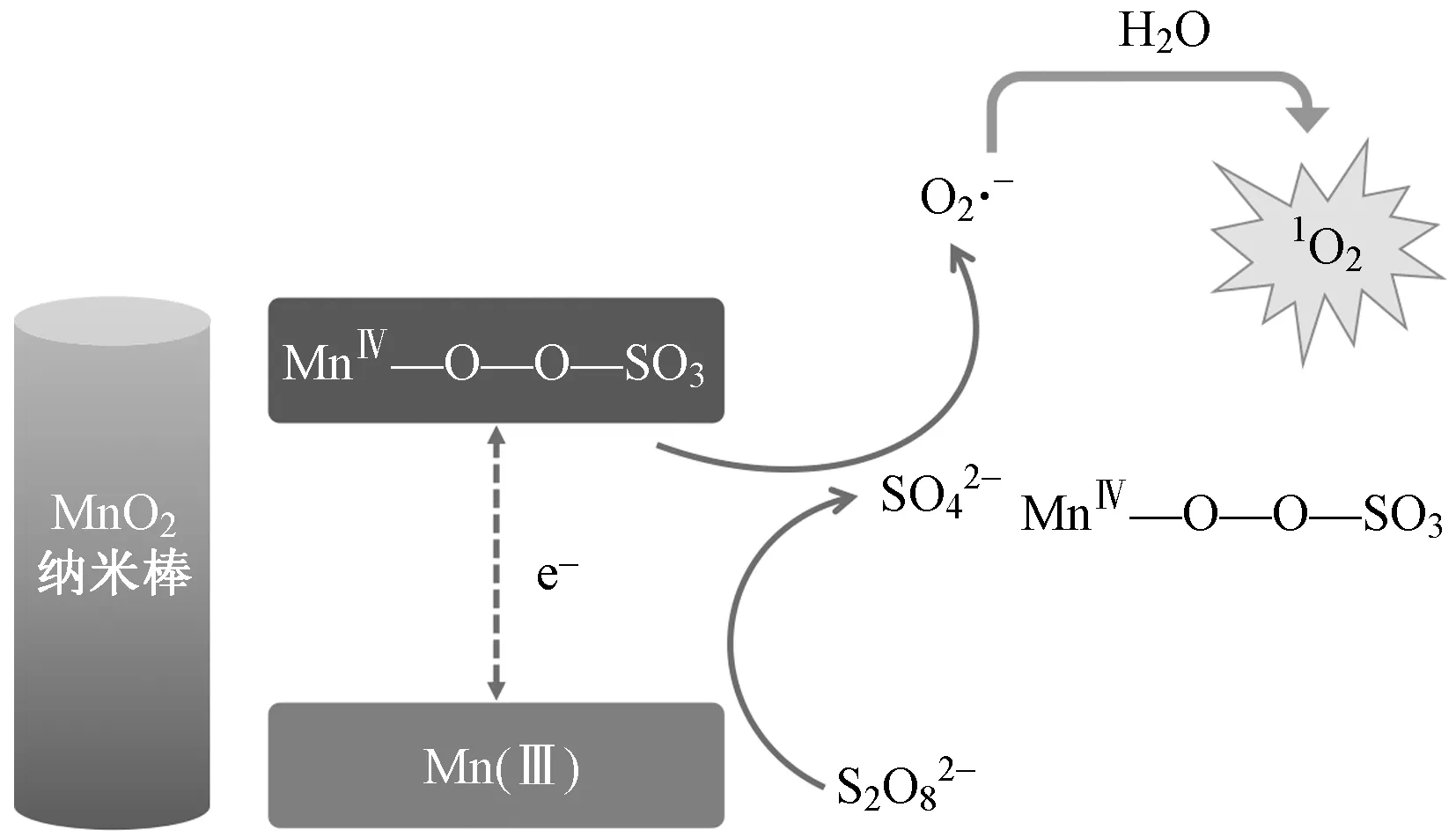
图2 MnO2纳米棒活化PDS生成单态氧的反应机理[80]Fig.2 The reaction mechanism of activation of PDS by MnO2 nanorods to generate singlet oxygen[80]
4.2 MnOx及其复合型氧化物活化PDS
除MnO2之外,MnOx(MnO、Mn3O4、Mn2O3和纳米零价Mn等[81])也可以用于活化PDS降解污染物〔见式(25)(26)〕. MnOx与其他金属氧化物复合能够有效提高催化反应活性.
(25)
(26)
Shah等[82]利用Mn0、Mn(Ⅱ)、Mn(Ⅲ)活化S2O82-生成SO4·-和·OH降解环丙沙星(CIP). 由于生成了反应性较弱的S2O8·-以及部分电子从S2O82-转移到金属离子表面,使Mn(Ⅲ)/S2O82-降解CIP的效率较低. Mn0可再次生成Mn(Ⅱ),从而保证Mn(Ⅱ)持续充足的供给. 并且Mn0的表面积较大,能够将目标污染物CIP吸附至表面从而提高催化活性. Li等[83]证明在3种Mn(Ⅲ)Ox中,γ-MnOOH相较于α-Mn2O3和Mn3O4在降解苯酚方面表现出更好的催化反应活性. 在中性和碱性条件下,SO4·-和·OH是降解苯酚的主要活性物质,而在酸性条件下苯酚的降解主要是γ-MnOOH和PDS反应形成的氧化中间体引起的. γ-Fe2O3/Mn3O4纳米复合材料表面Fe(Ⅲ)被还原成Fe(Ⅱ)后与S2O82-发生类Fenton反应,生成SO4·-和Fe(Ⅲ)[84]. 随着Mn3O4的引入,Mn(Ⅱ)充当电子转移体,促进Fe(Ⅱ)的再生,提高反应活性[85].
5 结论与展望
a) 锰基氧化物的合成存在多种方法,在选择有效的合成方法之前,要充分考虑锰基氧化物的表面结构、物理化学性质、浓度等因素. 通过改变反应时间、反应温度、溶液中Mn的总浓度等,能够调控锰氧化物的合成路径. 水热法是其中最常用方法之一,单独使用水热法合成锰基材料或与其他方法相结合,均可以制备具有较高催化反应活性的锰氧化物催化剂.
b) 水环境中存在的大量无机阴离子(HCO3-、NO3-、SO42-、CO32-等)与SO4·-或·OH反应,抑制水体中污染物的降解去除. 溶液的pH对反应性能的影响不显著,反应温度通常与有机污染物的降解率呈正相关,污染物的初始浓度与降解率呈负相关. 此外,晶型结构、表面积、氧化态也是影响锰基氧化物反应活性的重要因素.
c) 锰基氧化物活化PMS/PDS存在自由基(SO4·-和·OH)和非自由基(1O2)2种反应机理. 锰基氧化物活化PMS/PDS产生SO4·-,而·OH可由SO4·-与H2O或OH-反应生成. 同时,复合型锰基氧化物相较于单一MnO2具有更好的催化反应活性. MnO2活化PMS与PDS的反应机理不同:Mn(Ⅳ)被PMS还原生成SO5·-和Mn(Ⅲ),随后被氧化成Mn(Ⅳ)并生成SO4·-;活化PDS是Mn(Ⅳ)与Mn(Ⅲ)之间的氧化还原,先生成S2O8·-后产生SO4·-. 此外,MnO2活化PDS存在仅产生1O2的非自由基反应机理.
d) 相比于活化PMS,利用锰基氧化物活化PDS降解废水中有机污染物的研究较少,且二者的降解机制不同. 大多数锰基氧化物活化PMS过程通常会产生SO4·-和·OH,但β-MnO2活化PDS仅产生1O2. 未来需深入研究锰基氧化物活化PDS的机理.
e) 锰基氧化物经多次循环使用后仍具有较高的催化活性和稳定性. 但目前除了利用磁性对部分金属氧化物进行回收外,大多使用过滤法. 因此,研发高效、经济实用且易于快速分离的催化剂具有重要意义. 较多的复合型锰基氧化物催化剂已被证实具有较好的催化活性,但复合型氧化物催化剂的合成工艺较一元氧化物更为复杂. 在较温和的条件下合成高效的复合氧化物催化剂可作为未来研究方向之一.
猜你喜欢
化学与生物工程(2022年9期)2022-09-30
材料与冶金学报(2022年2期)2022-08-10
环境工程技术学报(2022年3期)2022-06-05
能源化工(2021年6期)2021-12-30
陶瓷学报(2021年4期)2021-10-14
科学与财富(2021年33期)2021-05-10
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2020年6期)2021-01-26
合成技术及应用(2021年1期)2021-01-07
作文成功之路·小学版(2020年6期)2020-07-27
