
细菌全基因组测序技术用于沙门氏菌流行病学调查的可行性分析
2022-06-07 05:51林本夫任欣悦袁晓琪梁梦诗丘穗萍潘婧淇原丽红
中国动物检疫 2022年6期
林本夫,任欣悦,袁晓琪,梁梦诗,丘穗萍,潘婧淇,原丽红
(1.广州市花都区动物卫生监督所,广东广州 510800;2.广东药科大学生命科学与生物制药学院,广东广州 510006)
食源性疾病一直是全球广泛关注的公共卫生问题之一[1]。据统计[2],每年有6亿人患食源性疾病,其中42 万人死亡,每10 人中就约有1 人患病。其中,由沙门氏菌(Salmonella)引起的食物中毒人数居我国食源性致病菌致病人数的首位[3]。当前沙门氏菌实验室诊断方法(GB 4789.4—2016)[4-5]成本高、周期长,对操作人员技术要求高,不能满足基层高通量、快速、低成本的检测需求[6]。全基因组测序(whole genome sequencing,WGS)以特异性基因差异来表征菌株之间的差异,具有显著的分型优越性[7],并且可以快速详尽地得到耐药细菌的特征,是检测细菌耐药性的有效技术手段,极大克服了传统方法的局限性[8-9]。
前期对分离到的9株沙门氏菌进行了传统的血清分型和耐药性分析[10]。本研究以这9株沙门氏菌为研究对象,通过全基因组测序预测其血清型和耐药基因,进而将全基因组测序结果与传统检测结果进行对比,评价两者结果的一致性和各自优缺点,探讨全基因组测序技术在沙门氏菌基层检测和监控工作中的应用前景。
1 材料与方法
1.1 材料
从猪、禽屠宰场环境样品中分离得到9株沙门氏菌,其中6株来自生猪屠宰场(土壤2株、污泥2株、屠宰车间拭纸2株),3株来自禽类屠宰场(土壤1株、污泥1株、屠宰车间拭纸1株),具体采样和分离方法见参考文献[10]。
1.2 方法
1.2.1 全基因组测序 9株沙门氏菌全基因组测序,由广东美格基因科技有限公司(中国广州)完成。提取细菌基因组DNA,对DNA 进行完整性和纯度检测;使用NEB Next UltraTMDNA Library Prep Kit(Illumina)进行建库和质检;利用Ilumina Novaseq 6000 平台对文库进行测序,生成150 bp的对端序列。
1.2.2 基因组数据质控、拼接和组装 对沙门氏菌基因组测序原始数据,采用FastQC 质控分析合格后,通过SPAdes_3.14.1(https://github.com/ablab/spades)进行拼接组装,去除<200 bp的重叠群片段;用RAST(rapid annotation using subsystem technology,https://rast.nmpdr.org/rast.cgi)统计沙门氏菌基因组大小、GC 含量等信息。
1.2.3 血清型预测和MLST 分型 根据全基因组测序结果,通过SeqSero 1.2(https://cge.cbs.dtu.dk/services/SeqSero/)进行血清型预测,同时在沙门氏菌基因组中检索7 个看家基因(aroC、dnaN、hemD、hisD、purE、sucA和thrA),将序列上传至MLST 2.0(https://cge.cbs.dtu.dk/services/MLST/)中,与数据库中原有的等位基因型序列进行比对,得到每个管家基因的编号,将其组合后得到对应的序列型(sequence type,ST)。
1.2.4 耐药基因分析 基于9株沙门氏菌的全基因组测序分析结果,采用ResFinder 3.2(https://cge.cbs.dtu.dk/services/ResFinder/)对菌株基因组所包含的耐药基因进行预测。耐药基因预测参数为ID >90%,且序列长度大于目的基因长度的60%;基因组组装中,去掉覆盖度<20 的节点。
1.2.5 与传统分析结果对比 将9株沙门氏菌全基因组测序结果与传统血清分型和耐药性检测结果进行对比。传统血清分型和耐药性检测,根据GB4789.4—2016 对样品进行增菌/选择性培养基培养、筛选,随后经PCR 扩增、测序鉴定,利用传统血清分型方法对沙门氏菌O/H 多价和单因子血清型鉴定;根据行标WS/T639—2018 选择庆大霉素(GEN)、氟苯尼考(FFC)、环丙沙星(CIP)、阿莫西林(AMX)、氨苄西林(AMP)、诺氟沙星(NFX)、头孢噻吩(CEF)、头孢噻肟(CTX)、头孢曲松(CRO)、头孢他啶(CAZ)、氨曲南(ATM)、亚胺培南(IPM)等12 种临床常用抗菌药物,对沙门氏菌分离株进行抗药性分析。具体检测方法及结果判定参考文献[10]。
2 结果
2.1 全基因组测序
9株沙门氏菌编号分别为P-SSL1、P-SSL2、P-SSL3、P-SSL4、P-SSO1、P-SSO3、A-SSL3、A-SSL1、A-SSL4,基因组总长度为4 738 737~5 161 611 bp,基因平均长度为41 964.3~103 016 bp,GC 含量平均为52.06%。其中:P-SSL1、P-SSL2 的GC 含量最少,为51.9%;P-SSL3、P-SSL4 的GC 含量最多,为52.2%;A-SSL1 大于200 bp 的重叠群片段数量最少,仅46 条,基因组长度也最短,仅4 738 737 bp;P-SSL2 大于200 bp 的重叠群片段数量最多,为123 条,基因组长度也最长,为5 161 611bp。具体结果见表1。
2.2 血清型预测和MLST 分型
9株沙门氏菌中共预测到3 种血清型和ST型,分别是鼠伤寒沙门氏菌(S.typhimurium,ST34)、德尔卑沙门氏菌(S.derby,ST40)、肠炎沙门氏菌(S.enteritidis,ST11),具体见表1。其中:鼠伤寒沙门氏菌为优势血清型,占55.56%(5/9),其次为德尔卑沙门氏菌和肠炎沙门氏菌,均占22.22%(2/9)。分离自猪屠宰场的6株沙门氏菌中,有5株鼠伤寒沙门氏菌、1株德尔卑沙门氏菌;来自禽屠宰场的3株沙门氏菌中,有2株肠炎沙门氏菌、1株为德尔卑沙门氏菌(表2)。
2.3 耐药基因预测
通过对9株沙门氏菌全基因组进行耐药基因分析,得到7 大类抗菌药物共22 种耐药基因,包括氨基糖苷类[aac(3)-IVa、aph(3′)-Ia、aph(4)-Ia、aadA1、aadA2、aadA3、strA、strB]、β-内酰胺类(blaTEM-1B、blaOXA-1)、氯霉素类(floR、catB3、cmlA1)、喹诺酮类[qnrS2、aac(6′)Ib-cr]、利福平(ARR-3)、磺胺类(sul1、sul2、sul3、dfrA12)、四环素类[tet(A)、tet(B)]。其中:氨基糖苷类耐药基因中携带率最高的是strA和strB基因,携带率均为66.67%(6/9);β-内酰胺类耐药基因中携带率最高的是blaTEM-1B基因,携带率为66.67%(6/9);磺胺类耐药基因中携带率最高的是sul2基因,携带率为88.89%(8/9);其余种类耐药基因携带率均不超过55.56%。在预测的所有耐药基因中,携带率最高的耐药基因是磺胺类的sul2基因(88.89%),携带率最低的是氨基糖苷类的aadA3基因(11.11%)。不同菌株间耐药基因差异较大,在22 种耐药基因中,有10 种耐药基因的检出率超过55.56%;9株沙门氏菌均携带3 种及以上的耐药基因。具体结果见表1。
2.4 与传统分析结果对比
利用传统血清分型方法共鉴定出鼠伤寒沙门氏菌、肠炎沙门氏菌、德尔卑沙门氏菌3 种血清型。其中分离自猪屠宰场的6株沙门氏菌中,有5株鼠伤寒沙门氏菌、1株德尔卑沙门氏菌;来自禽屠宰场的3株沙门氏菌中,有2株肠炎沙门氏菌、1株为德尔卑沙门氏菌。基于全基因组测序预测的血清分型结果与传统的血清分型结果[10]一致(表2)。

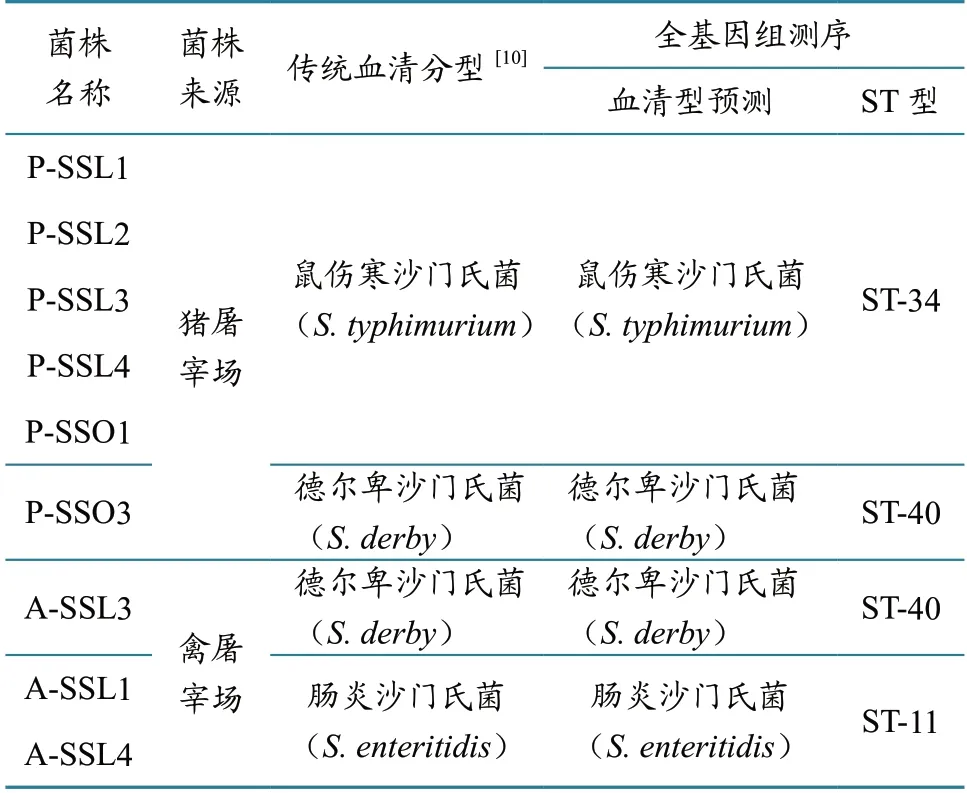
表2 基于全基因组测序的血清分型结果与传统血清分型结果对比
传统试验时选择了基层畜禽养殖实践中常用的4 类(β-内酰胺类、喹诺酮类、氨基糖苷类和氯霉素类)12 种抗菌药物,对9株沙门氏菌进行药敏试验(表3),发现9株沙门氏菌对阿莫西林和氨苄西林完全抗药,猪屠宰场分离到的6株沙门氏菌中有4株对庆大霉素耐药,有3株对氟苯尼考耐药;禽屠宰场分离到的3株沙门氏菌中有1株对庆大霉素、氟苯尼考和环丙沙星耐药。而基于细菌全基因组数据的耐药基因分析结果显示,共检测到7 大类22 种耐药基因,其中针对β-内酰胺类、喹诺酮类、氨基糖苷类和氯霉素4 类抗菌药物的检测结果与传统检测方法结果一致。全基因组测序与耐药性分析对比结果表明,β-内酰胺类、氯霉素类、喹诺酮类和氨基糖苷类药物的耐药表型[10]与耐药基因的检测结果符合率为100%,即所有耐药菌株中均检测到相应的耐药基因。此外,全基因组测序预测到利福平、磺胺类和四环素3 大类抗菌药物的7 个耐药基因,表明9株沙门氏菌可能对这3 大类抗菌药物耐药。

表3 基于全基因组测序的耐药基因分析与药敏实验结果对比
3 讨论
沙门氏菌作为主要的食源性病原菌,严重威胁着人类健康。由于沙门氏菌分型多且复杂[11],加之长期使用某种抗菌药物甚至滥用抗菌药物导致(多重)耐药菌株出现,尤其是在畜牧业养殖中,为防治疾病感染,促进动物生长,大量的抗菌药物被用作饲料添加剂,使得基层沙门氏菌病防控工作面临严峻挑战[12-13]。
对沙门氏菌进行快速准确的分型和耐药判断,针对性地指导临床用药,对于沙门氏菌病防控尤为重要[14]。临床上常用的传统血清分型方法是通过玻片凝集法确定O 抗原和H 抗原[15],然后根据沙门氏菌血清抗原表确定血清型。此方法对血清质量要求高,且花费大、用时长、人工成本高;对某些凝集不明显难以进行判别的情况依赖于技术人员的经验积累,容易造成误判;此外,某些罕见血清型由于缺少特异诊断血清,常常难以检测。相比之下,基于全基因组测序的分型方法,针对细菌特异性基因的差异来表征菌株之间的差异,用时短,准确率高,避免了常规血清分型中主观判断的影响,同时对传统血清学检测技术无法鉴定的菌株也可以进行分析,是一种高分辨率的分型方法。分子分型技术(如MLST、cqMLST、wgMLST 等)随着基因组时代的来临将成为当前细菌分型研究的热点[16]。
监测沙门氏菌耐药性,研究其耐药形成主要机制,对沙门氏菌病的预防和控制具有重要意义。沙门氏菌耐药性一般分为天然耐药和获得性耐药。天然耐药是细菌的固有特性和赖以生存的基本条件;获得性耐药主要是由于长期药物使用而逐渐形成,主要由可以转移的DNA(转座子或者质粒)介导,可以水平传播[17]。沙门氏菌耐药性种类的增多,加大了依赖抗菌药物治疗沙门氏菌病和基层沙门氏菌病防控工作的难度,因此在食源性动物饲养过程中抗菌药物的使用应严格限制并遵守休药期,以减少细菌耐药性的产生,保障食品安全和人类健康[18]。
实验室在进行耐药性分析时,主要基于当地抗菌药物使用情况选择性进行药敏分析,无法进行大规模药敏试验,且该方法培养周期长,易受培养基、培养条件、人员操作等因素的影响。全基因组测序准确率高,操作简便,用时更短,预测耐药基因范围更广,而且ResFinder 抗性基因数据库在预测耐药基因方面可以检出更多的耐药基因,是耐药性分析的首选工具。耐药基因对耐药性的预测也为明确耐药机制以及耐药性的检测提供了新的视角和方法[19]。当出现新的血清型或耐药基因时,可直接通过全基因组测序数据进行检索和分析,无需再次进行常规的细菌培养鉴定,为沙门氏菌血清型和耐药性的分析提供了更加简便的方法。
食品安全问题在我国乃至全球已经成为一个重要的公共卫生问题,食源性致病菌作为食品安全的一个主要因素逐渐受到人们的重视。为确保我国食品安全,保证人民健康,防止食源性感染的发生,需要开展广泛的流行病学调查,建立完善的食源性疾病监测网络[19]。全基因组测序技术已应用于细菌性传染病暴发调查和流行病学分析中,如2010年海地地震后的霍乱暴发菌株溯源,2011 年发生在欧洲的O104:H4 大肠埃希菌暴发事件调查,发生在加拿大持续3 年的结核病暴发分析,新生儿重症监护室耐甲氧西林金黄色葡萄球菌暴发的调查,耐碳青霉烯类抗菌药物肺炎克雷伯菌医院内感染暴发调查等[20]。此技术在沙门氏菌流行病学中为抗病、生长、食欲、代谢调节、表型、环境适应机制及重要经济性状基因的分析提供重要数据,促进了对畜禽遗传资源的深入研究与利用[21]。
随着全基因组测序的不断成熟,以及数据库的不断完善,会出现更多基因数据库和研究软件,为今后的研究提供了便捷[22]。基于全基因组测序的沙门氏菌分型和耐药性分析方法的检测成本和周期也将会进一步降低,测序速度、测序读长也将会进一步增加,细菌分型和耐药基因分析范围会更广,能够满足基层沙门氏菌病防控的高通量、快速、低成本的需求,从而指导沙门氏菌病防控的用药。因此,基于全基因组测序开发操作简单、低成本、高效、快速的沙门氏菌分型和耐药性分析方法,在沙门氏菌流行病学调查和基层疫病防控具有一定的应用价值,相信全基因组测序也会被广泛应用于更多物种的基因组研究。
猜你喜欢
动物医学进展(2022年9期)2022-09-06
世界科学技术-中医药现代化(2022年3期)2022-08-22
中国饲料(2021年17期)2021-11-02
医药前沿(2021年25期)2021-10-16
中国畜禽种业(2021年7期)2021-08-01
昆明医科大学学报(2021年5期)2021-07-22
祝您健康(2019年12期)2019-12-12
食品与生活(2015年12期)2015-12-14
智能制造(2015年4期)2015-05-12
江苏农业科学(2014年6期)2014-08-12
