
CO2 还原铜基电催化剂的原位重构效应研究进展
2023-01-31 04:48匡禹豪张文彪高庆生
复旦学报(自然科学版) 2022年6期
匡禹豪 ,张文彪 ,高庆生 ,唐 颐
(1.暨南大学 化学与材料科学学院,广东 广州 511400;2.复旦大学 化学系,上海 200433)
随着化石燃料的过度消耗,全球变暖问题已经引起了全世界的关注[1-3]。根据国际能源署(International Energy Agency,IEA)2018年的报告[4],全球的能源需求在2040年将增加30%,而温室气体二氧化碳(CO2)的排放量将达到357亿吨/年。在此背景下,电化学二氧化碳还原反应(CO2Reduction Reaction,CO2RR)成为了研究热点[5-8]。研究人员为了得到拥有更高活性和稳定性的催化剂,需要理解催化过程中的反应机理以及实际反应过程中催化剂活性位点的变化[9-10]。近些年来,多种原位测试技术得到发展并应用于CO2RR,可用于追踪/监测催化剂表界面的重构行为,实时分析电极表面的反应状态,为更深入认识实际反应条件下的催化过程提供了基础[9,11-12]。例如,在CO2RR还原反应初期,无论氧化铜(CuO)的初始状态如何,都会被还原成低价的铜物种(Cu0,Cu+),形成新的更稳定、活性更高的活性位点[13-15]。此外,电催化剂的重构行为在长时间的反应过程中同样会对活性位点产生正面或负面的影响。例如,金属铜(Cu)电极在电化学CO2RR过程中会经历溶解和再沉积的过程,导致催化剂表面结构发生变化从而失活[16-17]。因此,无论是对机理研究还是实际应用,深入认识并有效调控Cu基催化剂的电化学重构行为都有重大的意义。
本综述对CO2RR 中的Cu基电催化剂重构效应的相关研究进行了归纳和总结。首先,本文将Cu基催化剂的重构行为简单分为: 氧化态的改变和表面形貌结构的变化,详细介绍了相关研究进展,并介绍基于重构规律的电催化剂设计策略。然后,介绍研究催化剂重构效应的若干原位表征技术,归纳总结各自的优势、劣势。最后,基于上述进展,提出了电催化剂重构的一些展望,包括: 机器学习与实验观测相结合、催化剂稳定性提高、实用型电催化剂的发展等。
1 电化学二氧化碳还原反应(CO2 RR)的催化剂重构
1.1 CO2 RR
在所有的CO2转化技术中,由于不需要苛刻的反应条件(温度、气压等),电化学还原CO2具有广阔的应用前景[18-19]。如图1(a)所示,电化学CO2还原成燃料和化学品是间歇式储存能量的有效途径。图1(b)为电化学转化CO2的主要产物,有一氧化碳、甲酸、甲烷、甲醇、乙烯、聚碳酸酯衍生物和苯甲酸衍生物等,它们的价格从1 300美元/吨(一氧化碳、甲酸)到400美元/吨(甲醇)不等。众所周知,CO2RR 需要具有高活性的均相或多相催化剂。
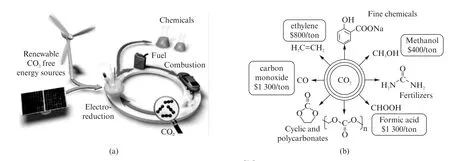
图1 (a) 用电化学方法将CO2 还原为燃料和工业化学物质[20],(b)CO2 还原产物中主要有价值的商品化学品[21]Fig.1 (a)Electrochemical reduction of CO2 to fuels and industrial chemicals[20],(b)Main valuable commodity chemicals from the CO2 RR[21]
多相催化剂上,CO2RR 通常发生在电极/电解质界面,其中电极是固体电催化剂,而电解质通常是CO2饱和的水溶液。如图2所示,催化过程包括3个主要步骤: 首先,CO2在电催化剂上发生化学吸附;随后,电子转移使C—O 键断裂或者质子迁移形成C—H 键;最后,吸附态的甲酸根(*COOH)既可以发生重排,从电极脱附至电解液,形成甲酸根离子(HCOO-),又可以与溶液中的质子反应脱水形成CO,还可以继续加氢形成烃类、醇类或者醛类[22-23]。其中,被广泛认可的是,活性物质是CO2自由基负离子,由CO2在电极表面吸附还原形成,自由基的产生是整个反应的第一步且是最重要的一步[24-26]。
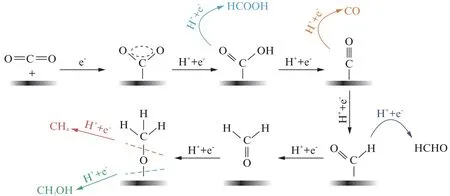
图2 中性溶液条件下电化学CO2 还原成5种C1 产物形成路径和机理[27]Fig.2 Reaction mechanism of electrochemical CO2 reduction on electrodes in aqueous solutions and the formation paths for the five main C1 products during the reduction process[27]
1.2 铜基CO2 RR电催化剂
铜是少数能将CO2还原成C2+产物(碳氢化合物和醇)的电催化剂。许多研究工作都致力于提高CO2在铜电极上电化学还原过程的效率和选择性[13-14,28-30]。虽然近些年来许多非铜基催化剂也表现出电催化CO2还原为C2+产物的能力,但产物得率较低[31]。因此,大部分的研究工作都关注不同结构铜基电催化剂的电催化性能,主要有: 金属铜[14]、铜合金[32]、铜基化合物(氧化物、磷化物等)[13,32]以及负载型的铜催化剂(Cu/C,Cu/SnO2)[33-34]等。由于CO2RR 需要高效的活性位点,因此确定催化剂的活性组分及其在反应过程中起作用的活性位至关重要。然而,催化剂往往随着反应的进行发生变化,这就给确认活性位点和设计高效的电催化剂带来了困难。同时,电化学反应过程中铜表面结构的原位重构和氧化物种的生成也会影响产物的选择性。因此,总结铜基催化剂的重构现象,以及原位观测催化剂反应过程中的尺寸/结构/形态变化就显得十分重要。
1.3 Cu基电催化剂的重构现象
在电场的作用下,催化剂的原位重构主要包括价态的变化和表面结构的变化。前者是在反应电位下电催化剂发生氧化或还原,并伴随有组成和相的变化。例如,在CO2RR 过程中,Cu的氧化物被还原为金属Cu物种[35]。后者强调表面纳米结构的重构,例如粗糙度、孔隙度、结晶度和晶面的改变。即使在没有发生氧化还原过程的情况下,表面原子和反应中间体之间的强相互作用也能改变催化剂的界面能,使催化剂发生原位重构[36]。例如,反应中间体*CO、*H 在Cu表面上的强吸附,容易导致催化剂发生原子重排和表面重构[37-38]。
原位重构可以基于热力学原理进行一些预测。例如CO2RR 往往在相对较负的电位下进行,容易驱动Cu氧化物或氢氧化物发生原位还原[39],这使得CO2RR 中铜基电催化剂的原位还原普遍存在[40]。然而,由于工作电催化剂固-液界面上反应物浓度的偏差[41-42],实际的热力学状态与标准热力学状态有很大的不同,催化剂与反应中间体的结合也会增加热力学预测的不确定性。因此,仅对催化剂原位重构进行热力学预测是不够的,需要引入动态现场原位表征,以监测电极微环境,从而监测催化剂的原位重构。
1.3.1 Cu基催化剂的氧化还原
由于CO2具有非常稳定的分子结构,需要非常高的能量才能活化。因此,CO2RR 往往需要非常负的工作电位,这使铜基氧化物/氢氧化物容易还原为金属铜。与纯金属铜相比,铜基氧化物/氢氧化物通常具有较好的性能,这表明原位形成的铜纳米颗粒上的活性位点起到了关键作用[43-44]。当用电化学抛光的金属铜作为电催化剂时,CO2RR 得到的产物高达16种[45],但是CO2转化的法拉第效率随着反应时间迅速下降;而利用铜衍生的氢氧/氧化物的电催化剂具有对C2+产物较高的法拉第效率(>50%)[46-48]。许多研究团队证明了在CO2RR 的反应条件下,氢氧/氧化物衍生的铜催化剂表面存在一定量的含氧物种,会增强C2+产物的选择性[13,16,49-50]。然而,在较高的还原电位下,Cu(OH)2/Cu O 会还原成金属铜。Lei等[13]制备了两种铜基催化剂HQ-Cu(含有Cu,Cu2O,Cu O)和AN-Cu(含有Cu,Cu(OH)2)。与电化学抛光的铜基底相比,HQ-Cu表面呈海绵状,而AN-Cu呈现纳米线的形貌(图3(a)~(c))。图3(d)中两种Cu电极的原位X 射线衍射(X-Ray D iffraction,XRD)图谱证明,HQ-Cu含有Cu、Cu2O、Cu O,AN-Cu含有Cu和Cu(OH)2。研究结果表明无论Cu的初始状态如何,在CO2RR 过程中电极都被还原为Cu(O),高的C2+选择性与初始的特定氧化状态无关,重构过程中产生的晶界增加和高指数晶面是C2+产物高选择性的原因(图3e)。该研究团队利用聚焦离子束从电极中提取超薄样品,通过电子显微镜和电子能量损失光谱研究不同Cu物种的分布和进化(图3(f)和(g)),结果表明这种氧化-还原的过程生成了能促进C—C耦合的晶界和高指数晶面,增强了氢氧/氧化铜催化剂对C2+产物的选择性。
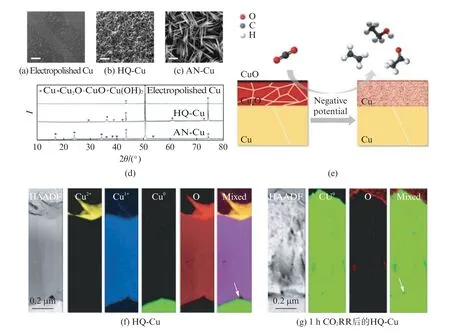
图3 SEM 图: (a) 电化学抛光Cu,(b)HQ-Cu,(c)AN-Cu;(d)3种铜电极的XRD;(e) 氧化铜电极上CO2 RR 示意图;(f) 高角环形暗场STEM;(g) 图(f)对应的EELS[13]Fig.3 SEMimages of(a)electropolishedCu,(b)HQ-Cu,and(c)AN-Cuinwhichthe scale bars all represent 2μm;(d)Indexed XRD patterns ofthesethreeelectrodes;(e)SchematicCO2RRonoxidized Cu-electrodes;High-angle annular dark-field(HAADF)STEM image and corresponding EELS mapping on different Cu oxidation statesand oxygen oftheFIB-fabricated specimen that corresponds tothe crosssectionof(f)HQCuand (g)HQ-Cu after 1 hofthe CO2RRat-1.05Vvs RHEin CO2-saturated 0.1mol/L KHCO3[13]
与上述研究相似,Grosse研究团队发现在不同尺寸的Cu 立方体上,Cu Ox经CO2RR 后还原为Cu(0)[51]。碳负载的Cu立方体上含量为52%的Cu2O 在CO2RR 后几乎完全还原为Cu(0),仅残留4.6%的Cu(I)。尽管Cu Ox在CO2RR 过程中被还原为Cu(0),但残留的Cu(I)物种往往是不可避免的。因此在CO2RR 过程中,铜表面的氧化态分布需要特别注意。Aràn-Ais等[52]发现在CO2RR 的过程中,Cu(I)很难被完全还原。Jung等[47]指出当撤掉施加的电压后,原位还原生成的Cu纳米颗粒容易再次氧化。根据Chou等[53]的研究结论,这种铜表面的氧化态分布可能与CO2RR 机理的变化有关。他们通过电沉积的方法制备了铜催化剂代表Cu(I)表面,未处理铜催化剂代表Cu(0)表面,CV 处理过的铜催化剂(在CO2RR 条件下经100次CV 处理循环)代表Cu(I)/Cu(0)表面,得到了完全不同的产物分布(图4(a))。CV处理过的铜产生更多烃类产物(C2H4和CH4的法拉第效率分别为40%和35%)。他们通过原位表面增强红外吸收光谱(SEIRAs)研究了Cu表面不同的CO 种类(图4(b))。桥式吸附CO(CObridge)相关峰的强度在~30 min达到最大值,而线式吸附的CO(COatop)峰在前15 min时增加,然后随时间的推移减小。由于X射线吸收光谱(X-ray Absorption Spectroscopy,XAS)在前15 min主要观测到Cu(I)的特征峰,并且Cu(I)在60 min内完成到Cu(0)的转变。如图4(d)所示,COatop来自富含Cu(I)的电沉积Cu表面,能进一步还原为CH4等C1产物。但在富含Cu(0)的表面生成CObridge,这抑制了烃的生成。CV 处理的Cu电极有不同的Cu(I)/Cu(0),有利于COatop和CObridge共存,因此对CO2还原生成乙烯具有较高的选择性。通过多种策略可以协同提高CO2RR 反应催化剂的性能,包括调节电解液的状态、物理性质以及铜催化剂的氧化态。Eilert等[28]利用原位X 射线吸收光谱证明了铜立方体在CO2RR 过程中具有很高的活性,而且对乙烯的选择性很高,实验结果表明形成铜立方体的前驱体是铜的氧化物,而不是实验过程加入的物料CuCl2(图5(a~d))。针对铜的氧化物,Chou等[49]通过原位表面增强红外吸收光谱(SEIRAS)观测铜表面在CO2RR 过程中的氧化态分布,证明了线性吸附COatop中间物种的形成是在Cu(I)表面,而桥式吸附CObridge是在Cu(0)表面(图5(e))。
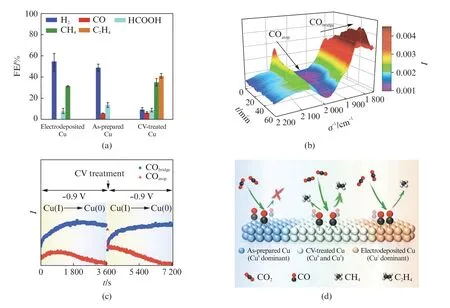
图4 不同含量Cu(0)和Cu(I)的铜表面上的CO2 RRFig.4 CO2 RR on a Cu surface containing different amounts of Cu(0)and Cu(I)
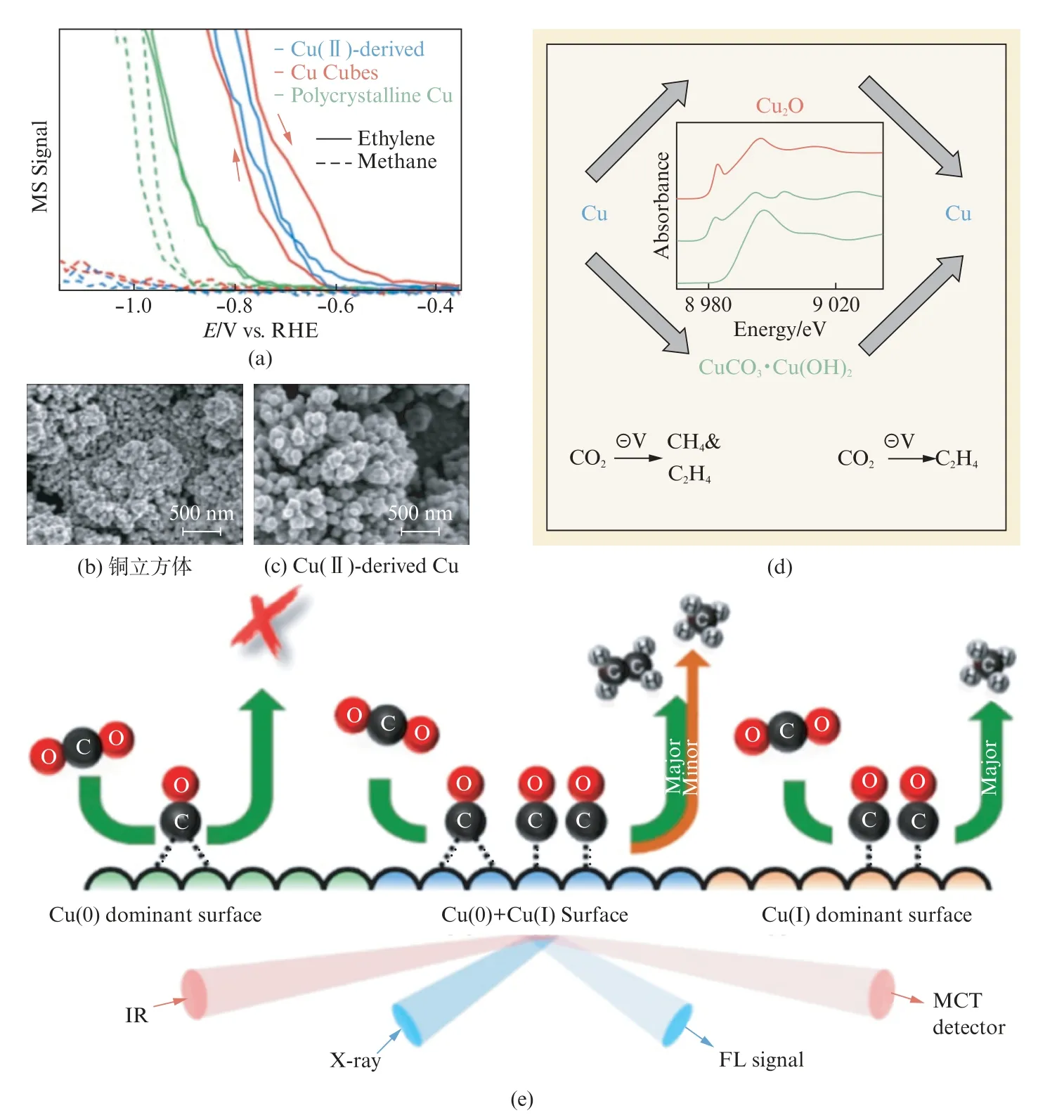
图5 (a) 不同铜催化剂CO2 RR 过程中的OLEMS[28];(b) 铜立方体和(c)Cu(II)-CO3/OH 的SEM 图;(d) 铜催化剂形成示意图;(e) 铜表面CO2 RR 过程示意图[49]Fig.5 (a)OLEMS during CO2 RR while cycling the potential using different Cu catalysts;(b)SEM images of Cu cubes and(c)Cu(II)-carbonate/hydroxide derived electrocatalyst;(d)Schematic of formation of copper catalysts;(e)Schematic illustration of the electrochemical CO2 reduction on the Cu surface[49]
双金属铜基催化剂,如载体型铜基催化剂和合金催化剂,同样受到关注[32-33,54]。例如Cu/SnO2的复合电催化剂,包括异质结构、核壳结构等,该催化剂在H 型电解池中对CO 产物表现出良好的催化性能[54]。包信和研究团队设计了一种新型的Sn-Cu催化剂,该催化剂具有高导电性的分等级结构:Sn-Cu合金/Sn金属核和无定形薄SnOx壳,表现出极高的催化性能[33,54]。原位的X 射线吸收精细结构(Extended X-ray Absorption Fine Structure,EXAFS)谱(图6(a~b))和非原位的X 射线光电子能谱(Xray Photoeletron Spectroscopy,XPS)及透射电子显微镜(Transmission Electron Microscope,TEM)照片(图6(c~f))表明: 在CO2RR 的还原电位下,SnOx壳可以原位重构形成Sn/SnOx界面。这种分等级结构中的Sn核可以提供足够的Sn源,使得Sn扩散到壳内,加速重构。在该工作中,催化剂壳体的表面结构可以通过核体的组成精确控制,这为铜基多金属催化剂的设计提供了新思路。
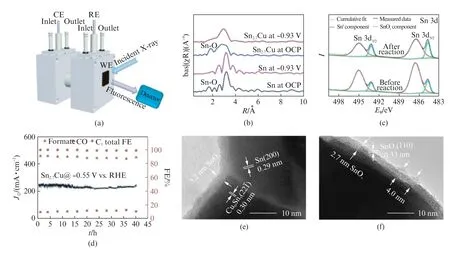
图6 (a)CO2 RR 原位EXAFS装置示意图;(b)Sn和Sn2.7 Cu催化剂在-0.93 V vs.RHE原位Sn K-edge EXAFS;(c)Sn 3d XPS;(d)CO2 RR 稳定性测试;反应前(e)以及反应后(f)的TEM 图[33]Fig.6 (a)Diagram of in situ EXAFS liquid cell for CO2 RR;(b)In situ Sn K-edge EXAFS spectra of the Sn and Sn2.7 Cu catalysts operated at open circuit potential(OCP)and -0.93 V vs.RHE;(c)Sn 3d XPS spectra of Sn2.7 Cu catalyst before and after CO2 RR stability test;(d)CO2 RR stability test in flow cell of Sn2.7 Cu.(e)Ex situ HRTEM image of Sn2.7 Cu catalyst before and(f)after CO2 RR stability test[33]
1.3.2 铜基催化剂的表面重建
近年来的研究发现,铜催化剂的活性受高指数晶面的暴露和不饱和配位的铜原子影响[12,34,55-56],因此表面形貌结构对活性起到关键作用。与催化剂的体相结构相比,表面结构更加不稳定。相关研究表明Cu催化剂在CO2RR 过程中的表面重构会导致纳米颗粒的形貌发生变化,对催化剂的活性和稳定性也有一定的影响[16-17,57-58]。例如,Osowiecki等[59]研究了铜纳米晶在CO2RR 过程的动态演化,结果表明Cu纳米晶在CO2RR 过程重构的影响因素包括CO2浓度、颗粒负载量以及溶液pH 等,而中间体*CO 是导致Cu表面形貌变化的关键因素,能引起颗粒尺寸、暴露晶面、聚集状态等的变化。Kim 等[57]利用多晶铜电极在一个固定的还原电位下进行CO2RR 反应,发现了Cu电极经历逐步表面重构,在30 min内先形成Cu(111),再经30 min后形成Cu(100),而在Cu(100)表面形成后没有发生进一步的表面转变。这一结果有助于解释多晶铜在CO2RR 反应中的产物选择性,同时也为通过廉价的多晶铜电极来生产铜(100)单晶电极提供思路。
铜纳米催化剂的形貌可以通过退火、电沉积等方法控制,在CO2RR 中对多碳产物的选择性有显著的影响。除此之外,由于电位较高,在电解过程中铜会溶解并重新沉积,导致表面发生形貌变化而其氧化态不变。Huang等[37]通过研究3种不同大小的纳米铜立方体在电催化CO2RR 过程中的变化,阐述了结构与活性、选择性和稳定性的关系。图7(a~c)(见第676页)显示3种尺寸的铜纳米颗粒在反应过程中都经历了纳米团簇形成,然后纳米团簇聚集的过程。而在实际CO2RR条件下,(100)、(110)和(111)晶面的规整程度随着电势的减小而减小,其中更负的电势倾向于形成稳定非立方粒子,这是由于吸附物覆盖的铜(111)和(110)表面的表面能更小(图7(d))。如图7(e)所示,第一阶段是铜溶出再沉积重新形成纳米团簇(StageⅠ),随着纳米团簇的形成,纳米铜立方体结构开始受到破坏(StageⅡ)。这些结果表明,整个纳米颗粒的重构可能更倾向于形成小颗粒,这是因为小颗粒具有更高的表面能以及小颗粒聚集成大颗粒的过程更快。

图7 (a~c)3种尺寸Cu NCs的TEM 随时间变化图;(d)pH 与电位对Cu纳米颗粒的Wulff形状的影响;(e)Cu NCs在CO2 RR 过程中的变化机理[37]Fig.7 (a-c)Representative Cu NCs of three different sizes:a 16 nm,b 41 nm,and c 65 nm,imaged with TEM at different operation times.The rectangle in a encloses an aggregated assembly of particles.Thered and yellow arrows in b indicatesmallclusters and brokenCu NCs,respectively.Scale bars:100nm;(d)pHand potential dependence of the Wulff-shape of Cu nanoparticles including H,CO,and mixed H+CO covered surfaces;(e)Overview of the degradation mechanism of CuNCs during CO2 RR.Schematic representation of the degradation mechanism that includes nanoclustering(StageⅠ)followed by a coalescence at a later stage(Stage Ⅱ)[37]
最近的研究报道了溴化铜枝晶可以催化CO2RR,选择性产生乙烯,法拉第效率达到57%,这主要归因于高曲率的形貌以及不饱和配位的金属位点[60]。De Luna等[16]从溶胶-凝胶前驱体中电沉积合成铜催化剂,该方法减缓了铜的电化学还原过程,使得纳米结构得到控制,并且Cu+能够在较低的电位下稳定。随着电化学还原电位的不同,铜催化剂出现了不同的结构形态。在-0.7 V vs.RHE 时,表面形成了一圈0.5~5μm 的纳米结构,而在-1.0 V vs.RHE时尖锐的纳米枝晶开始生长。在-0.7 V时,与前驱体相关的峰都消失了,但出现了Cu2O和金属Cu的峰。在更负电位时,Cu对应的峰值开始增加,而Cu2O 峰值消失。Cu+到Cu0的转变相对于Cu2+到Cu+的转变速度更慢,这个过程可以同时控制形态和氧化状态。原位X射线吸收光谱对CO2还原条件下铜氧化状态随时间变化的研究结果证明了高电位下Cu+的存在。
Jung等[47]最近的工作表明,晶面的控制可能不是提高C2和C3产物法拉第效率的唯一方法,C2H4选择性的增加还与反应过程中催化剂形态转变有关。在-1.1 V 的电位下,20 nm 立方Cu2O 晶体颗粒经历原位电化学反应破碎成2~4 nm 小颗粒(图8(a~c)),碎裂会从纳米晶体的表面开始。与Cu@Cu O/C或块状Cu箔相比,被破坏成颗粒状的催化剂可提高C2+选择性,并且抑制了H2的产生。原位X 射线吸收近边结构研究在CO2RR 过程中观察到Cu Ox的形成,但尺寸小的纳米颗粒更多在电解质内部的开路电势下被重新氧化,从而产生不稳定的Cu状态。
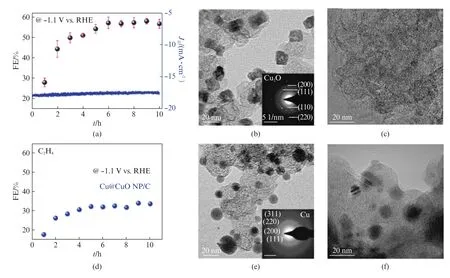
图8 (a)Cu2O NP/C中C2 H4 的法拉第效率;(b)CO2 RR 前和(c)CO2 RR 后的Cu2O NP/C TEM;(d)Cu@CuO NP/C生产C2 H4 的法拉第效率;(e)CO2 RR 前和(f)CO2 RR 后Cu@CuO NP/C的TEM[47]Fig.8 (a)Faradaic efficiency for C2 H4 production for Cu2O NP/C;TEM of Cu2O NP/C(b)before and(c)after the CO2 RR test;(d)Faradaic efficiency for C2 H4 production for Cu@Cu O NP/C;TEM of Cu@Cu O NP/C(e)before and(f)after the CO2 RR test[47]
关于Cu原位重构的机理研究表明,CO2RR 过程中Cu催化剂表面结构的原位重构可能是由电迁移效应和电化学效应引起的[37-38,59,61]。前者是在负电位的驱动下,电流流动导致固体金属物质发生质量传递[62],克服表面原子迁移的动力学势垒。后者是表面Cu原子在CO2RR 过程的中间体(如*CO、*H、*OH等)的强相互作用下发生原位重构。负电位和中间体的强相互作用能够克服表面重构的动力学势垒,甚至改变原有的热力学关系。
在Kim 等[57]的研究中,多晶铜在CO2RR 过程中先重构形成Cu(111),之后重构形成Cu(100)。其中,多晶铜转变为Cu(111),符合热力学预测结果,而Cu(111)重构形成Cu(100)则与热力学预测结果不符,这可能是在高负电位下静电斥力驱动表面原子重构为表面不密集的Cu(100)结构。最近,Lee等[38]用原位掠入射X 射线吸收光谱(GIXAS)和原位掠入射X 射线衍射(GIXRD)表征了CO2RR 条件下多晶铜薄膜的近表面结构,发现只有在CO2存在的条件下,多晶铜才会重构为Cu(100)。Huang等[37]的理论研究指出,*H 或*CO 的吸附改变了Cu(111)、(100)和(110)面的界面自由能,导致了在CO2RR 工作电位下的重构。
1.4 基于重构现象设计CO2 RR电催化剂
了解催化剂重构的机制有助于利用重构形成性能更好的活性相。研究人员在铜基催化剂改进方面已经取得了一定的进展,如稳定电解过程中产生的活性位点或构建活性物质。高价态铜在CO2RR 中的还原现象普遍存在,通过诱导还原可将铜的氧化物、硫化物、氮化物、卤化物等重构,提高铜催化剂的电催化性能。Jiang等[63]用密度泛函理论研究了不同Cu表面对CO2RR 的影响,在铜箔上合成了Cu2O(100)表面的纳米立方体,Cu2O 原位还原为保持立方结构的Cu并优先暴露Cu(100)。与抛光的铜箔相比,最佳的Cu纳米立方体催化剂C2+与C1的产物比提高了6倍,C2+的法拉第效率超过60%,相应的电流密度超过40 mA·cm-2。
表面结构是铜基催化材料开发的关键,对催化剂性能有着关键影响。例如,Kim 等[64]通过铜纳米颗粒转化得到的铜立方结构,可以在较低电位下将CO2电化学还原为多碳产物。铜纳米颗粒在电化学还原条件下的结构演化可能是促进其多碳产物生成的关键。该团队探究了先前报道的铜纳米颗粒在CO2还原条件下的结构转变过程[65],表明一个原位形成的无定形铜结构可能是C2+活性提高的关键,然而这种无定形结构会迅速氧化,导致活性下降。因此,可采用化学钝化技术来阻止这种氧化。
2 原位表征技术在研究CO2 RR 电催化剂重构效应中的应用
在对CO2RR 电催化剂的研究中,基于不同表征技术进行的原位研究是重要的手段,设计更先进的原位表征技术对深入研究催化反应有着重要意义[66-70]。对反应进程中催化剂的原位结构进行详细的讨论,有助于识别真实的活性位点,为揭示反应机理提供思路。
原位XRD 技术是基于布拉格衍射的一种重要的固体物理表征方法[71]。通过分析得到的随时间变化的衍射图,可以揭示催化剂在反应过程中的结晶或非晶化行为。由于材料的衍射强度取决于原子的类型和位置,原位XRD 可以检测出催化剂的单位晶胞的大小、形状和取向。采用原位XRD 技术实时检测催化剂的晶相,可以进一步分析催化剂的稳定性和相变,从而为更好地设计高效催化剂提供技术支持。值得注意的是,原位XRD 技术只能用于分析结晶样品,不适合结晶度差的样品;同时,由于空间分辨率较低,无法检测到局部位点或组分。
另一种检测催化剂重构的技术是原位拉曼光谱(Raman spectroscopy)。拉曼光谱具有较高的空间分辨率(<1 nm),适用于检测微观催化剂。通过对拉曼光谱的分析,可以得到被测物质的振动和旋转能级,从而对物质进行识别。原位拉曼光谱通常用于评估表面物质的价键变化,有时用于检测电解质中的产物[72-73]。然而,由于拉曼光谱是基于分子键的振动,因此拉曼光谱对纯金属不敏感。
与拉曼光谱不同的是,XPS可以用于深度探测电催化剂,而不限定于催化界面。激发光电子的能量只与原子轨道有关,它能定性地确定元素类型和价键或成键模式[74-75]。此外,原位XPS技术可表征催化过程中活性位点的电子转移行为,这对进一步揭示催化反应机理具有重要意义。原位XPS技术只能探测到近表面范围的元素,不能揭示催化剂内部结构的变化。
作为独特的X 射线技术,原位同步辐射XAS探测X 射线辐射的非弹性散射的能量从几百到几千电子伏,可以监测内部的电子跃迁和目标原子的化学状态,分辨出掺杂物和主元素的状态[9,76-77]。XAS主要测试元素的化学状态和电子结构,而真正的空间分布、原子的成键条件和配位环境可以通过EXAFS谱的拟合结果推断得到[78]。原位XAS技术虽然重要,但只能描述局部结构的电子/配位环境,无法得到催化剂宏观的信息。
不同于上述所提到的技术,原位TEM 作为一种精密的测量技术,可以实时观察催化剂在反应过程中的形态变化。然而,到目前为止,原位TEM 技术在电催化领域的应用还处于起步阶段[79]。TEM 实验中电子激发源的能量对于一些结构敏感的样品来说过高,而原位TEM 实验的工作环境和样品制备相对严格,特别是对于高分辨率的测量。因此,研究人员使用透射电镜非原位观察形态变化,而不需要了解电子结构和化学状态,这应该是揭示形貌结构演变相对较好的手段。
总的来说,原位表征技术为实时检测催化反应的动态演变提供了可能,进一步加深了对催化机理的认识。
3 总结与展望
由于提供了生产高价值化学品且同时回收碳资源的新方案,利用可再生能源电催化CO2还原反应已经引起了广泛的关注。近年来发现了许多引人注目的纳米结构与反应活性和选择性的关系。然而,为了使该工艺更接近商业和应用水平,还需要进一步了解催化剂的内部性质。本文对CO2RR 电催化剂的原位重构进行了综述,突出了以往研究中被低估的这一关键问题。更加深入的研究将进一步揭示局部原子规模效应,强化重构催化剂在现实应用中的可行性。
此外,目前电催化剂原位重构的进展局限于现象观察,还没有对催化过程中催化剂的演化进行系统的理论研究。大部分已有的理论研究目的是讨论催化剂表面上反应物与产物之间的关系,而不是“催化剂本身的变化”。研究重构效应的最终目的是通过对自重构过程的有效跟踪,建立预催化剂、自重构过程和真实催化剂之间的系统理论,进而揭示催化剂原位重构的内在机理,预测催化剂重构的方向。特别是对于高过电位和酸性/碱性电解液等反应条件下阴极还原过程的动态演化,实验技术难以实现原位表征。但通过控制计算条件和优化反应参数,机器学习可以模拟整个反应过程,预测电催化还原反应的动态演化,从理论分析中推断重构机理。将原位研究与机器学习相结合是未来电催化研究的一个重要方向,有望大大提高研究效率。
最后,CO2RR 反应作为碳循环的一个基础反应,其催化机理的研究已经取得一定进展,但催化剂在此过程中的变化决定了其稳定性,同时也是催化剂能否广泛应用的关键。因此,认识CO2RR 电催化剂的重构效应对提高催化剂稳定性,设计实际应用的催化反应系统具有指导意义。
猜你喜欢
实用手外科杂志(2022年2期)2022-08-31
哈尔滨工业大学学报(2022年5期)2022-04-19
摄影世界(2022年1期)2022-01-21
陶瓷学报(2021年5期)2021-11-22
中国生殖健康(2020年7期)2020-12-10
天然产物研究与开发(2018年5期)2018-06-13
山东工业技术(2017年21期)2017-11-04
商周刊(2017年6期)2017-08-22
浙江大学学报(工学版)(2016年2期)2016-06-05
火炸药学报(2014年1期)2014-03-20
