
二硫化钼加氢脱硫催化剂研究进展
2023-01-31 04:48闫天兰闫玥儿张亚红
复旦学报(自然科学版) 2022年6期
闫天兰 ,闫玥儿 ,张亚红 ,唐 颐
(1.复旦大学 化学系,上海 200438;2.复旦大学 图书馆 中华古籍保护研究院,上海 200433)
随着世界原油重质化和劣质化加剧以及环保要求的提高,市场对轻质清洁油品需求急剧增加[1]。但重油中较高含量含硫化合物的存在,导致石油炼制过程中催化剂中毒、反应设备腐蚀等问题,且含硫燃料在使用过程中释放出硫氧化物,会引起酸雨等一系列环境污染,甚至危害人体生命健康。因此,世界各国对燃油硫含量的要求越来越苛刻,我国已全面实施的国VI标准要求车用燃油硫含量不大于10 mg/kg。目前,油品脱硫技术主要有萃取脱硫[2]、吸附脱硫[3]、氧化脱硫[4]和加氢脱硫[5]。其中萃取脱硫条件温和、能耗低,但脱硫能力受溶剂影响较大,对油品普适性差;吸附脱硫操作条件温和、吸附效率高,但吸附剂存在吸附容量低、再生频繁的问题;氧化脱硫工艺简单,但需要额外使用氧化剂且砜的分离过程繁琐;加氢脱硫虽然所需条件苛刻,氢气和高温高压设备成本较高,但其对油品适用范围广、脱硫性能好等优势是其他脱硫方法无法比拟的。因此,加氢脱硫是实现超深度脱硫最具潜力的技术,也已广泛应用于国内外轻质油品的生产。
为实现油品低硫化甚至零硫化的目标,开发价廉易得的高活性加氢脱硫催化剂逐渐成为炼油化工领域的焦点[6]。过渡金属硫化物特别是二硫化钼(MoS2)具有独特的层状结构,由于其活性高、稳定性好和成本低等优点在加氢脱硫领域已经有很长的发展历史,被广泛用于原油加氢脱硫及超低硫清洁油品的生产[7]。人们已经对其制备方法和反应机理做了较为详细的总结,如Zhang等[8]和Mao等[9]总结了MoS2制备方法和应用研究的现状,Lauritsen等[10]综述了不同含硫分子在MoS2模型催化剂上的吸附位点及催化机理,Tanimu等[11]综述了包括MoS2在内的加氢脱硫催化剂的制备及反应机理。
近年来,伴随着电催化水分解制氢、生物质加氢脱氧等应用的快速发展[12-13],人们对MoS2催化剂的活性位结构有了更精准的表征,提出了新的调制方法及其构效关系认识,这些新认识有可能对加氢脱硫催化剂的设计、改进及应用性能的提升具有重大指导作用。因此,本文将进一步试图总结近年来MoS2催化剂在构筑方法、活性相模型及作用机制等方面的研究进展,重点阐述MoS2加氢脱硫催化剂的活性相结构及其催化性能的构效关系。
1 MoS2 加氢脱硫催化剂的构筑方法
1.1 MoS2 基本结构
MoS2是典型的层状过渡金属硫化物,基本结构如图1(a)所示。单层MoS2由3层原子层构成,上下两层为硫原子层、中间一层为金属钼原子层,形成S-Mo-S的“三明治”夹心结构,多层MoS2由单层MoS2通过层间微弱范德华力结合而成[14]。
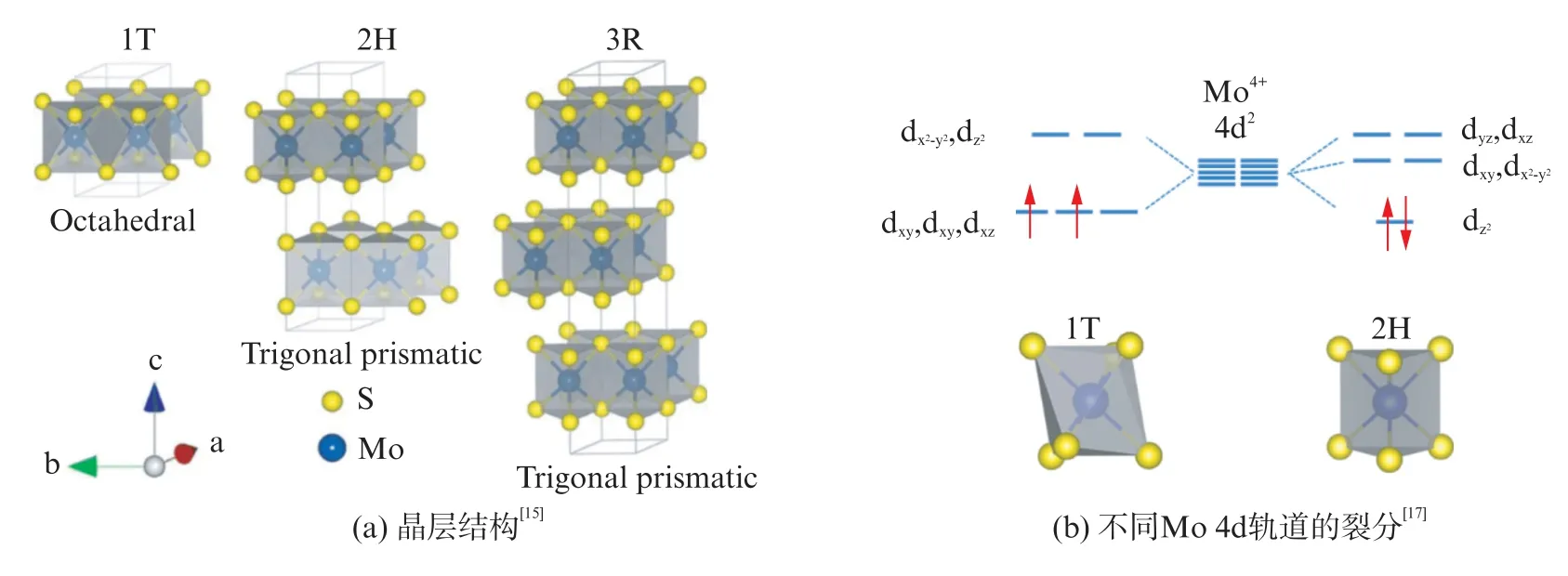
图1 MoS2 的基本结构(1T、2H 和3R 相)Fig.1 Structure of 1T,2H and 3R MoS2 polytypes
根据S-Mo-S不同的配位结构以及堆积方式,MoS2可分为1 T、2 H 和3R 相[15]。2 H-MoS2的每个晶层内Mo原子位于S原子形成的三棱柱中心,与周围的6个S原子配位,并沿c轴以每两个S-Mo-S晶层为一个结构单元重复堆叠而成,具有六方晶系的配位结构。与2 H 相不同,1 T-MoS2的晶层内上下两层S原子呈不对称排列,以2 H 相中三棱柱结构为基础,将其中一层S原子绕顶面中心旋转60°所得,Mo原子与周围的6个S原子呈八面体配位,并以这种具有特殊S-Mo-S结构的晶层沿c轴重复堆叠。3R-MoS2具有与2 H-MoS2相同的原子配位结构的晶层,但是其晶层沿c轴排布形式与2 H 相不同,以每3个S-Mo-S晶层为周期排布,因其较为罕见故不作详细介绍。1 T 和3R构型均属于热力学亚稳态,2 H-MoS2属于热力学最稳定状态。1T 和3R 构型均可在一定条件下转化为2 H,因此2 H-MoS2的研究最为广泛。
根据晶体场理论,MoS2中Mo原子外层价电子排布为[Kr]4d55s1。其中4个价电子与配位S原子形成共价键,其余2个价电子填充在Mo 4d轨道中[16-17]。由于Mo-S配位对称性不同,导致Mo 4d轨道裂分不同(图1(b))。在1T-MoS2中,MoS6八面体配位结构使Mo 4d轨道裂分为t2g和eg两组轨道,其中较低能级的t2g为4dxy、4dyz和4dxz组成的三重简并轨道,两个价电子分别占据其中的两个轨道,使得1TMoS2在Fermi能级附近存在未填满的空轨道,从而表现出类金属性[18]。而2 H-MoS2中,三棱柱配位的MoS6结构使Mo 4d轨道裂分为和3组轨道,两个价电子以自旋相反的形式填满能级最低的由所构成的轨道,因此表现出半导体性质。
基于MoS2特殊的晶层结构及其堆叠特点,现阶段研究主要集中于调控MoS2的晶层结构、尺寸和层数来调控MoS2的形貌和电子结构,进而调控其催化性能。
1.2 MoS2 晶层的构筑方法
1.2.1 2H-MoS2晶层的构筑方法
2H-MoS2合成方法主要包括水/溶剂热法、化学气相沉积法、高温硫化/分解法和电化学法等[19]。
1) 水/溶剂热法
水/溶剂热法通常在高温、高压的密闭反应釜中进行,且制备条件至关重要[20]。Luo等[21]探究了制备条件对MoS2形貌的影响,发现随着水热温度和时间的增加,MoS2从珊瑚状的聚集颗粒变为了花状的球形再到包裹的纳米片形貌。Lee等[22]以四硫代钼酸铵(ATTM)为前体水热制备了MoS2,并探究了pH值对MoS2形貌的影响,发现随着pH 值增加,MoS2形貌从不规则颗粒聚集体变为纳米薄片堆积的花状团簇。Xiong等[23]利用盐酸调控溶液中硫离子的浓度,通过一步水热法成功合成了高度分散的MoS2纳米花,如图2(a)所示。Zhang等[24]和Lu等[25]分别以聚乙烯吡咯烷酮(PVP)和聚吡咯为模板水热合成了MoS2纳米花(图2(b)和(c))。Leng等[26]研究了添加不同醇类对水热合成MoS2形貌的影响,发现醇类(乙醇、乙二醇和甘油)辅助合成的MoS2纳米花尺寸更小、层间距更大。
2) 化学气相沉积法
化学气相沉积法(CVD)是指钼源和硫源在高温、等离子体、真空或紫外线等特殊条件下发生化学反应,在衬底上直接生长MoS2。段司航等[27]调整了CVD 的工艺参数,制备出形貌不同、结构均匀、厚度可控的MoS2薄膜,并发现MoS2的形成受气相前驱体MoO3的扩散控制。Waduge等[28]通过优化CVD 工艺条件,在固态孔上成功制备了高质量、机械性能优越的MoS2薄膜(图2(d))。

图2 2 H-MoS2 晶层构筑的水热法(a~c)[23-25]与CVD 法(d)[28]Fig.2 The fabrication of 2 H-MoS2 layered crystals using hydrothermal method(a c)[23-25]and chemical vapour deposition(d)[28]
3) 高温硫化法
Yao等[29]在高浓度硫蒸气中硫化Mo O3制备了颗粒状MoS2。用惰性气体作为保护气,将Mo O3在600℃左右加热一段时间后和硫源混合进行硫化,可以制备出层状MoS2。Mo的氧化态对硫化度有一定影响,有效生成MoS2的前提是Mo在硫化前必须保持最高的Mo6+状态,而还原相MoOx的存在阻碍了MoS2的生长[30]。此外,Farag[31]发现硫化温度对MoS2结晶性影响较大,高结晶度的2 H 型MoS2需要硫化温度为800℃,在低于800℃的温度下硫化时会不同程度地出现Mo O2相。
4) 高温分解法
利用前体热解生成MoS2可避免催化剂硫化不完全等问题,是目前比较常用的方法[32]。一般通过控制热解温度、时间和前驱体S/Mo比等参数调节MoS2的尺寸以及形貌。
5) 其他
Jia等[33]以PVP为表面活性剂,采用一种简便可行的回流法合成了分支形貌的MoS2纳米线。PVP的引入是形成分支MoS2纳米线的关键,否则只能获得MoS2纳米颗粒。Zuo等[34]通过一种简便的气泡模板辅助法合成直径约100 nm 的具有富勒烯结构的MoS2纳米笼。此外,有研究人员开发出一种液-液界面制备MoS2的新方法,即在室温下不需搅拌或添加表面活性剂,通过有机溶剂中Mo分子前体的分解制备。
1.2.2 1T-MoS2晶层的构筑方法
近年来,除了三棱柱配位的2 H-MoS2,正八面体配位的[MoS6]所构成的1T 相和具有Mo-Mo成键的畸变1T 相(1T′、1T″和1T‴等)结构也逐渐受到科学家的广泛关注[36-37]。金属相MoS2是亚稳态的,在自然界中不存在,主要采用物理化学方法制备。到目前为止,已开发出十几种1T-MoS2的合成方法,可分为直接合成法和间接合成法两大类。其中,直接法一般以廉价的钼源和硫源为原料,在一定条件下反应,原位生成1T-MoS2,间接法通常以多层的体相2 H-MoS2为原料,经由相转化反应制备1T-MoS2。
1) 直接合成法
2016年,Geng等[38]首次报道了一种水热法合成稳定的1T-MoS2纳米片的方法,作者将其高稳定性归因于纳米片的高纯度和表面两侧吸附的单层水分子的存在(图3(a))。此前,纯相且稳定的1T-MoS2还不能通过化学方法合成出来。Takahashi等[39]在超临界水热条件下,以抗坏血酸为还原剂反应12 min成功制备出少层1T-MoS2。
2) 间接合成方法
该类方法以多层2H-MoS2为前体,利用层状MoS2层间间距较大且相互作用力弱的特性,采用层间嵌入碱金属、有机分子和3d过渡金属等方式制备1T-MoS2。插层过程中具有电子供体性质的插层剂进入层间,使得Mo的d带电子状态改变[40]。Fan等[41]通过类似的方法成功制备了金属相的MoS2纳米片,如图3b所示。但该方法存在产率低、锂化反应时间长等问题。针对上述问题,Wang等[42]借鉴锂离子插层的思想,利用电化学方法控制锂离子插层,成功制备了1T-MoS2(图3(c)),但该方法制备得到的单层1T-MoS2或1T-MoS2量子点产量低、面积小。随后,研究者从插层剂着手,Park等[43]采用熔融金属辅助插层的方法合成了1T-MoS2(图3(d))。Er等[44]发现NaK 合金是一种比Li更合适有效的插层剂,以Na K合金为插层剂可制备高1T相比例(94.5%)的MoS2。Zheng等[45]以萘基钠为插层剂,采用两步插MoS2。在上述研究基础上,Acerce等[46]利用电化学方法实现1T-MoS2层间不同离子(H+、Li+、Na+、层方法有效解决了插层离子扩散慢的问题,从而提高产量,成功制备了面积高达400μm2的单层1TK+等)的有效嵌入。然而,金属原子的过度插层会不可避免地破坏S-Mo-S 晶格结构[47]。随后,Peng等[48]采用直接化学剥离法首次成功合成高纯相(超过97%)、大尺寸的单层1T′-MoS2。

图3 1T-MoS2 晶层构筑的直接合成法(a)[38]和间接合成法(b)~(d)[41-43]Fig.3 The fabrication methods of 1T-MoS2 layered crystals using direct synthesis method (a)[38]and indirect synthesis method(b-d)[41-43]
此外,MoS2的1T 与2H 可以相互转变。早期Sandoval等[49]研究发现,化学剥离制备的单层MoS2在水悬浮液中呈现扭曲的八面体结构。随着水的排出和层内S-S相互作用力的恢复,单分子层重新堆叠,12天后结构就转为三棱柱配位的2H-MoS2。MoS2的相变机制是基面S原子的横向运动引起的,而S原子转移的驱动力是施加在1T-MoS2上的热能[50-51]。基于上述认识,有研究者直接以单层2H-MoS2作为前驱体,采用电子束、氩离子刻蚀等方法将单层2H-MoS2的三棱柱晶格原位转化为1T 的八面体晶格,从而获得1T-MoS2。Zhang等[52]发现碘化钾溶液处理可诱导单层MoS2从2H 相到1T 相的转变。Chang等[53]采用简单锂熔盐的策略,实现了单层2H 和1T-MoS2的精准合成。除此之外,杂原子掺杂也可以引起Mo与S之间配位结构的变化,从而获得1T-MoS2。
1.2.3 1T/2H 异质结构MoS2晶层的构筑方法
采用水热法、化学剥离法均可制备含有1T 相不同形貌的MoS2[54-55]。Li等[56]采用一步水热法合成了具有1T/2H 异质结构的MoS2纳米微球,通过简单地改变原料Mo/S的摩尔比,可有效调节MoS2的1T/2H 相比例。Liu等[57]发现改变Mo前驱体种类或水热温度也可实现1T 相比例的调控。Salimi等[58]研究发现在水热法制备MoS2的过程中,硫源种类对MoS2形貌和1T相比例影响较大,以硫脲为硫源得到纳米花状MoS2,而硫代乙酰胺为硫源可得到更高1T相比例的MoS2纳米片。Sun等[59]采用水热法制备抗坏血酸插层的MoS2,将其在Ar保护条件下经高温焙烧后得到高1T 相的MoS2纳米花,如图4(a)(见第688页)所示。Wang等[60]和Wu等[61]在水热过程中引入铵类前驱体,发现铵分解产生的小分子和离子很容易进入层间诱导1T/2H-MoS2的形成。添加有机胺N,N 二甲基甲酰胺(DMF)也可制备1T/2HMoS2,通过改变DMF与水的比例可以调控1T 相浓度[62]。Mohammadpour等[63]以糖基天然共晶溶剂为插层剂也实现了1T/2 H-MoS2纳米片的制备,如图4(b)所示。Luxa等[64]以碱金属萘盐剥离MoS2,发现碱金属阳离子半径是影响MoS2剥离程度和1T/2H 相比例的主要因素,如图4(c)所示。Zhang等[65]以溴化正丁基吡啶鎓(BPy)为插层剂(图4(d)),采用简便有效的水热法,利用BPy中较大位阻的Br和π堆积相互作用诱导MoS2从2H 到1T 相变,成功制备出高1T 比例(91.9%)的1T/2H-MoS2。Zhu等[66]利用具有一定动能的氩离子轰击单层MoS2表面,成功实现2 H 到1T 相的可控局域相变(图4(e))。借助扫描隧道显微镜,他们验证了具有直接带隙的2H 相和金属性的1T 相同时存在,并确认了极少量硫空位的存在使得这种1T/2 H 相共存的马赛克结构能达到稳定。此外,采用过渡金属掺杂的方法也可构筑异质结构的MoS2。

图4 1T/2H-MoS2 晶层构筑的水热法(a)[59]、化学剥离法(b~d)[63-65]与Ar离子轰击法(e)[66]Fig.4 The fabrication methods of 1T/2H-MoS2 layered crystals using hydrothermal method (a) [59],chemical exfoliation method(b-d) [63-65]and Ar plasma treatment (e) [66]
1.3 MoS2 层板大小调控方法
一般通过改变制备条件或合成方法来实现对MoS2尺寸的有效调控。屠智韫等[67]提出了常压CVD双管系统制备大尺寸均匀单层MoS2的方法,其尺寸大小在套管径向分布均匀,而沿轴远离钼源的方向逐渐减小。Tian等[68]以水热和微波辅助法分别制备了两种MoS2,其中通过微波辅助法获得了20~30 nm粒径均匀的小尺寸MoS2,而水热法得到的MoS2尺寸较大。
1.4 MoS2 层数调控方法
MoS2层数的调控方法主要有物理剥离法、化学液相剥离法、CVD 和原子沉积法(ALD)[69]。由于MoS2层间通过较弱的范德华力相连接,因此通过胶带可轻易地从块状材料中剥离出纳米级多层或单层MoS2材料[70]。Liu等[71]对传统手撕胶带工艺进行改进,提出金膜胶带法,实现了大规模、高质量、大尺寸的单层二维MoS2晶体材料的制备。Yin等[72]采用溶剂热法对MoS2进行表面改性处理,实现了在低沸点极性溶剂中液相剥离制备少层MoS2。陈奎等[73]采用超声液相剥离法实现了3层结构MoS2的制备。Wang等[74]开发了液氮剥离法制备单层或寡层2H-MoS2纳米片。王子玄等[75]借助载气精准控制MoS2生长,成功获得单层MoS2,采用ALD 也可实现对MoS2层数精确调控[76-77]。
此外有研究表明,在超临界水热条件下通过改变有机还原剂种类即可实现MoS2纳米片层数调控[78]。使用强还原性的甲酸可制备出多层2 H-MoS2,而使用弱还原性的草酸则得到少层2 H-MoS2。也有研究发现延长水热时间和控制弱酸性条件都有利于制备高堆积层的MoS2催化剂[79]。
1.5 MoS2 层板大小和层数同步调控方法
CVD法可直接在衬底上生长面积与厚度可控的MoS2[80]。张鑫等[81]通过常压CVD 法制备了单层、双层及多层二维MoS2,并发现生长时间是影响二维MoS2层数的决定因素,生长温度则显著影响MoS2的尺寸。Yin等[82]开发了一种简便离心辅助液相剥离法,以多层大尺寸MoS2为原料,成功获得了具有纳米级横向尺寸、单层为主的MoS2纳米片。
值得注意的是,在催化剂设计过程中需根据催化反应的实际需求来调控催化剂的结构特征。为了获得催化性能优异的MoS2,不应仅局限于对MoS2相、层数及层板尺寸单一的调控。许多研究工作结合两种甚至多种方法优势,制备出合适层板尺寸、层数的MoS2。Tan等[83]结合球磨和化学插层法制备了超小尺寸、高1T 相比例的单层MoS2纳米点。
本质上MoS2的生长受动力学和热力学共同控制。基于对MoS2基本结构单元、合成方法的认识,理性调控Mo-S相互作用、晶粒的边界能和表面能以及生长过程的驱动力,可以实现MoS2的可控合成。
2 MoS2 活性相模型及活性相作用机制
催化剂的反应活性和选择性与其活性相结构密切相关,深入研究催化剂的反应活性中心结构和特点,了解其催化作用机理,有助于设计开发活性更高的MoS2基加氢脱硫(HDS)催化剂。为了阐明MoS2基催化剂的构效关系,学者们已经提出许多活性相理论模型(见图5),如rim-edge、corner-edge、硫空位和brim 理论等[84-85]。对HDS反应而言,目前普遍认为有两种反应途径,一种是直接脱硫路径(DDS),另一种是加氢脱硫路径(HYD),其反应活性和比例也与催化剂活性中心密切相关,因此有必要对其活性中心研究进展进行综述。
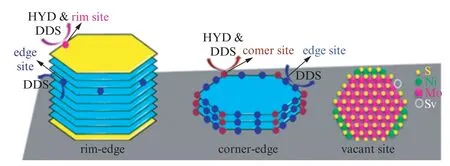
图5 MoS2 活性相模型:rim-edge、corner-edge、硫空位[84]Fig.5 Structure-activity models of MoS2:rim-edge model,corner-edge model,and vacant site model[84]
2.1 MoS2 活性相模型
(1)Rim-edge模型
1994年Daage和Chianelli提出rim-edge模型[86]。他们认为在MoS2中,存在具有加氢活性的rim位和氢解活性的edge位两类活性中心,其中MoS2片层的最上和最下晶层的边缘为rim 位,具有DDS和HYD活性,而中间晶层边缘为edge位,仅具有DDS活性。Rim-edge模型揭示了反应路径选择性与层数的关系,但该模型不能解释空间位阻小的噻吩HDS选择性变化原因。
(2)Corner-edge模型
考虑到小分子含硫化合物在MoS2层间没有空间位阻,有研究者提出corner-edge模型,如图5(b)所示,认为每个MoS2片层存在corner和edge两种活性位,由于corner位的配位不饱和度高于edge位,使得corner位的HYD 活性较高,故选择性随层板大小而改变[87]。
(3) 硫空位理论
一般认为边缘不饱和配位的硫空位Mo(CUS)是DDS路径的活性位。早期Kabe等[88]利用35S示踪法研究了HDS反应机理,认为催化剂上存在的不稳定S会在H2作用下生成—SH 和H2S,H2S脱附后产生硫空位和Mo3+。含硫化合物通过S原子吸附在硫空位处,发生加氢反应和C—S断键反应,留在催化剂上的S原子可再次在H2的作用下形成硫空位,完成整个循环。
(4)brim 理论
基于扫描隧道显微镜等精细表征手段,最近又提出MoS2的brim 理论。Lauritsen等[84]提出brim 位为MoS2完全硫化的Mo边,在STM 图中处于边缘明亮位置,具有较强的提供和接受电子的能力。虽然brim 位的Mo原子配位完全饱和,但容易吸附反应物分子,且附近Mo边的硫容易形成—SH,为加氢提供充足H 源,故认为brim 位是HYD 路径的加氢活性位。
以上模型从不同角度认识MoS2活性相,这些模型理论相互补充完善,虽然有不足或与其他理论矛盾的地方,但为MoS2催化剂对HDS反应的构效关系问题提供了新的见解。
2.2 吸附位点和反应过程
2.2.1 吸附位点的识别
现代科技水平的提升使研究者们可以直接监测HDS活性的相结构、表面吸附和反应路径,为开发高活性催化剂提供了更多的理论依据。
规划水资源论证是一项多学科交叉、专业性较强的工作,对从业人员的要求较高。目前,规划水资源论证缺乏专业技术人才,从业人员的技术水平有待提高。我国从事规划水资源论证的工作人员主要是长期从事建设项目水资源论证的持证上岗人员。论证人员在开展工作之前对规划水资源论证了解不多,未经培训就要承担规划水资源论证工作,往往会受到传统建设项目水资源论证思维的影响,从建设项目的角度思考问题。
早在2000年,丹麦科学家们通过扫描隧道显微镜STM 对MoS2模型催化剂进行表征,首次得到了单层MoS2纳米颗粒形状和边缘结构的STM 图像,发现完全配位饱和的MoS2呈三角形,且三条边均为Mo边。同时,作者也观察了边缘具有硫空位的MoS2的STM 图像[89]。随后研究发现在还原硫化条件下,MoS2为六边形[90]。Lauritsen等[91]进一步研究发现三角形MoS2纳米团簇的边缘结构取决于团簇的尺寸,n(n表示三角形MoS2边缘的Mo原子数)大于6的MoS2纳米团簇终止边是完全硫化的Mo边,而随着尺寸减小(n<6),MoS2终止边则转变为S边。
Lauritsen等[89]首先发现噻吩(C4H4S)可在单层MoS2纳米团簇的完全硫化边缘吸附并反应。Tuxen等[92]通过STM 研究了MoS2团簇上硫空位对含硫分子的吸附性能,发现并不是所有硫空位都能吸附,只有角位的硫空位才会吸附二苯并噻吩(DBT)。DBT 的加氢和脱硫分别发生在brim 位和角位。空间位阻较大的4,6-二甲基二苯并噻吩(4,6-DMDBT)在两类硫空位上都不吸附,而是平躺吸附在靠近MoS2团簇的brim 位。
2.2.2 反应路径
2004年,Lauritsen等[85]利用STM 揭示了噻吩在MoS2纳米团簇上的加氢脱硫反应路径。噻吩容易吸附在brim 位,当边缘存在—SH 时,噻吩加氢开环生成硫醇中间产物,随后该中间体很容易地转移到旁边硫空位处发生C—S键断裂反应,完成噻吩脱硫过程。Moses等[93]通过DFT 计算研究了噻吩在MoS2上HYD 和DDS的竞争反应,发现Mo边brim 位有利于噻吩的吸附,氢转移和加氢反应具有较低能垒,在S边上反应需要先形成S空位,C—S键的断裂更易发生在S边的空位。Zheng等[94]采用DFT 计算探究了噻吩在MoS2不同边上硫空位的HDS反应,发现DDS倾向于发生在Mo边,而HYD 倾向于发生在S边,Mo边的硫空位更有利于DDS路径的中间体和产物形成,由于丁烷的形成能垒较高,两边的主要脱硫产物均为丁烯。
Todorova等[95]研究了MoS2上的CH3SH 脱硫反应,发现CH3SH 通过S原子吸附在表面后S—H键先断裂,随后CH3基团结合相邻S—H 中的H 原子形成CH4完成脱硫。Mom 等[96]使用专用高压STM 原位监测模型催化剂上CH3SH 的脱硫过程,发现MoS2纳米岛活性边缘硫、氢和烃的覆盖情况随气体环境的变化而变化。在H2氛围中,即使痕量的S存在也会使边缘Mo原子被S的覆盖。对于高活性的MoS2催化剂,在加氢脱硫反应过程中,含硫化合物C—S键断裂生成的沉积硫会提高催化剂边缘S覆盖率。此外,反应速率或机理的细微变化可能对MoS2的边缘结构影响较大。因此,MoS2的活性位结构取决于催化剂的初始结构和原料性质。作者认为CH3SH 在MoS2上脱硫过程包括:1)CH3SH 转化为CH4;2)S原子以吸附态形式遗留在MoS2边缘;3) 硫原子在氢气作用下以H2S形式脱附3个步骤,其中CH3SH 的转化本质上是不可逆反应,而H2、H2S和CH3SH 的吸附/解吸过程都是可逆过程。
3 MoS2 结构对加氢脱硫性能的影响
由于1T-MoS2是热力学上的亚稳态结构,其近期发展主要体现在电化学储能以及电催化制氢等领域[97],而在高温高压下的HDS反应则罕见报道,因此后续主要基于2 H-MoS2相催化剂进行讨论。
3.1 MoS2 层数对催化性能的影响
Sun等[102]用有机胺代替无机铵盐水热制备了插层疏水纳米MoS2,有机胺的插入扩大了纳米MoS2的层间距,促进了稳定单层疏水结构MoS2的形成。该方法合成的疏水MoS2,具有更低空间位阻和更高边缘位点,表现出更优异的加氢脱硫性能和更高的加氢路径选择性。
3.2 MoS2 层板大小对催化性能的影响
Chowdari等[103]利用一种低温无模板法制备了MoS2纳米管,以NiMo氧化物为前驱体,改变硫化方法来调控MoS2纳米管的形貌。采用气相低压非原位硫化法可得到多壁MoS2纳米管,而使用液相高压原位硫化法则得到MoS2纳米砖沿垂直外表面自组装形成的MoS2纳米管。考察了两种催化剂对DBT加氢脱硫的催化性能,发现层板短、层数少的MoS2纳米砖初始反应速率提高了4倍。基于实验结果,作者建立了MoS2纳米颗粒尺寸与其活性之间的数学关系式,并对实验结果进行了验证。
在关注催化剂活性的同时,选择合适的HDS路径也至关重要。文献一般用HDS选择性因子SF,即含硫化合物的转化率与烃类加氢转化率的比值来表示路径选择性[104]。Zhang等[105]发现MoS2层板长度的增加显著提升了HDS路径选择性(SF从5.5增加至11.0),这主要是由于MoS2层板长度增加提高了edge/corner比,有效地降低了烯烃HYD 活性位。Li等[106]发现随着Co/MoS2±x中S/Mo原子比的降低,SF增加了两倍(从37提高到89),较低S/Mo原子比的Co/MoS2±x具有较高的HDS选择性,这是由于尺寸较大的MoS2暴露出更高比例的edge位。上述研究中MoS2构效关系均符合corner-edge模型,这些工作为调控和认识MoS2催化剂的HDS选择性提供了一种新思路。Zhang等[107]通过简单调节pH和前驱体类型,采用水热法成功制备了不同形貌的MoS2催化剂。实验结果表明: 随着MoS2层数增加,SF 从1.46增加到1.52,而层数相同、层板更长的MoS2则表现出更高的HDS选择性(SF从2.90提高到2.99),与层数相比,板长对MoS2的HDS选择性影响更大。有研究以四烷基硫钼酸铵为前驱体,通过原位热解制备了具有独特形貌孔结构的MoS2催化剂[108],其中四庚基硫代钼酸铵形成的MoS2催化剂直接断裂C—S键的选择性最高。
此外,配位不饱和点的低配位催化活性中心可能比基面上的原子活跃几个数量级,在这些位点上催化反应选择性也不同。因此,精准控制纳米团簇表面暴露的低配位位点是改善催化活性和选择性的一个重要方法[108]。Tuxen等[110]研究发现当MoS2纳米团簇的尺寸超过1.5 nm 时,与氢反应后主要形成边缘硫空位,但这些硫空位不能直接与DBT 键合。相比之下,小于1.5 nm 的MoS2纳米团簇的性能要好得多,硫空位主要在角位形成,对DBT 具有更高的亲和力。从这个角度讲,尺寸更小的MoS2纳米团簇可能具有对超深度脱硫制备清洁燃料独特的催化性能。
尽管早期许多研究表明MoS2的边缘位是HDS反应的优良活性位点。然而,相比于MoS2基面而言,边缘原子的数量较少。近期研究表明,2H-MoS2的基面并非惰性的,经改性后的基面也可提供活性位点[111]。因此,激发MoS2基面的大量原子参与催化反应是提高其整体催化活性的重要研究内容。目前的优化方法主要是通过在MoS2基面上构造原子空位或者掺杂异原子来提高其基面催化性能。Xu等[112]首次在单层MoS2上成功构筑弗兰克尔原子缺陷结构,并通过球差校正电镜确认其原子构型,结合理论计算和催化性能测试,表明不同缺陷结构会直接影响二维材料基面上的电荷分布情况,进而直接决定其催化活性。Hu和Zhou等[113-114]研究发现MoS2具有边缘硫空位和面内硫空位两种催化位点,面内硫空位是CO2高选择性加氢到甲醇的催化活性中心。但在加氢脱硫领域对MoS2这两种空位的具体作用机理尚未有深入研究。
3.3 1T/2H 异质结构MoS2 对催化性能的影响
近期报道也提出了以异质结构的1T/2 H MoS2为HDS催化剂。Wei等[115]以绿色无毒的乙醇/水为溶剂一步溶剂热法合成了纳米球状MoS2催化剂,该方法所制备的具有较高比例1T 相的少层MoS2展现了最佳的加氢脱硫催化性能。MoS2催化剂稳定性测试表明,DBT 转化率先略有下降后保持不变,而联苯(BP)的选择性先急剧下降后趋于稳定。随后,有研究学者利用一锅溶剂热法制备了基面1T-2H 相共生,且S和Mo空位共存的MoS2纳米花,通过精确控制溶剂热条件获得了具有高活性和高加氢选择性、富缺陷的1T-2 H MoS2催化剂。该催化剂在3次循环使用后仍然保持高性能[116]。最近,Bai等[117]在乙醇-水溶剂热体系中,通过对草酸的调控获得了1T相含量高、S和Mo空位适中的1T-2H MoS2纳米花,将其用于HDS反应。在循环实验中,MoS2催化剂的活性保持良好,且当催化剂第二次使用时,DBT 转化率高达98.3%,比第一次提高9%。此外通过上述3个例子还发现,在第一次反应中MoS2的结构还可能发生重构,如1T相比例降低、形貌变化等,对其深入研究将有助于更深刻认识MoS2催化剂的活性位点。
上述3项工作对于制备含有较高比例1T 相、层数可控或富缺陷且HDS催化性能良好的MoS2催化剂具有重要的指导意义。
4 小结与展望
随着表征技术的发展,科研工作者对MoS2在HDS构效关系理解方面已经形成一些认识并建立了相关活性相模型,为深入认识和理性设计高性能MoS2催化剂提供了新思路。但目前对MoS2催化剂的构效关系理解仍然有限,许多模型建立都是以单层MoS2为研究对象建立起来的,在催化反应条件下对实际MoS2原子尺度结构变化的原位解析仍有很大的挑战性,虽然其基面与边缘硫空位作用机制在电化学、CO2转化及加氢脱氧领域已经有了长足发展,但是对于高温高压下HDS反应领域的研究仍然匮乏。因此,深入认识MoS2结构、精准调控活性相结构及揭示其在加氢脱硫中的作用机制是MoS2加氢脱硫研究的重点。
猜你喜欢
分子催化(2022年1期)2022-11-02
物理学报(2022年17期)2022-09-14
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
电子制作(2019年15期)2019-08-27
表面工程与再制造(2019年6期)2019-08-24
陶瓷学报(2019年5期)2019-01-12
读者欣赏(2014年6期)2014-07-03
郑州大学学报(工学版)(2014年6期)2014-03-01
