
SLC6A1基因变异导致神经发育障碍的家系分析
2023-08-10 05:08陈丽卿付俊一苏堂枫
中风与神经疾病杂志 2023年4期
陈丽卿, 付俊一, 苏堂枫, 刘 艳
SLC6A1基因(MIM 137165)定位于染色体3p25.3,在人类和其他动物的发育期与成熟大脑中均广泛表达。SLC6A1基因编码γ-氨基丁酸转运蛋白1(gamma-aminobutyric transporter 1,GAT-1),该蛋白负责突触γ-氨基丁酸(gamma-aminobutyric,GABA)的再摄取和细胞外间隙清除[1,2]。当SLC6A1基因发生致病性突变时,GAT-1功能丧失,影响突触前膜从细胞外再摄取GABA,从而诱发癫痫。目前国内SLC6A1基因变异导致的癫痫报道少,主要报道的癫痫发作类型为肌阵挛-失张力发作及失神发作,且都为散发病例[3,4],我们报道1组SLC6A1 基因杂合变异引起癫痫的家系。
1 对象与方法
1.1 对象 先证者,女,于2岁6月龄就诊于我科门诊,系第1胎第1产,足月顺产,出生无窒息史。4月龄抬头,8月龄独坐,1岁8月龄独行。独立行走后出现腿抖,伴有频繁眨眼,独站不稳及摔跤,无腿抖时可以独行。就诊时会说2个字。于2岁3月龄时曾行康复训练,仍有间断腿抖,摔跤,不敢行走。语言无进步。查体:体重12 kg,头围49 cm。面容无特殊,四肢肌力、肌张力正常,病理反射未引出。指鼻试验不配合。可扶行,叫名无回应。血肝肾功能、电解质、乳酸、丙酮酸、血氨、空腹血糖、血尿代谢筛查正常。视觉及听力诱发电位正常。视频脑电图(video electroencephalogram,VEEG):背景节律慢化(δ、θ混合慢波);睡/醒期见双侧前头部、后头部或广泛性高波幅慢波、棘慢波或节律发放(见图1)。头部磁共振(magnetic resonance imaging,MRI):双侧额叶斑点状高T2Flair信号灶,少许脱髓鞘病变可能(见图2);左侧颞角较右侧大。Gesell婴幼儿发育量表:一般发育水平约21个月,各项发育落后,发育商64。儿童孤独症评定量表提示无明显孤独症表现。

图1 VEEG:背景节律慢化(δ、θ混合慢波);睡/醒期见双侧前头部、后头部或广泛性高波幅慢波、棘慢波或节律发放

图2 头部MRI:双侧额叶斑点状高T2 Flair信号灶,少许脱髓鞘病变可能
家族史:基因结果出来后追问病史获知患儿母亲年幼时有癫痫发作,曾口服药物治疗1年(具体不详)。目前无发作,但反应稍迟钝。患儿外婆幼时有抽搐病史,用药不详,目前无发作。
1.2 方 法
1.2.1 家系全外显子组测序 经监护人签署知情同意书,并经医院伦理委员会批准后,抽取患儿及父母,外婆外周血3 ml,提取DNA。应用 xGen Exome Research Panel v1.0 进行目标区域捕获,使用illumina Hiseq X-ten 二代测序仪对富集的文库进行 PE150 测序,平均测序深度为 100X,95%以上的区域达到 20X 以上的覆盖度。
1.2.2 多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)检测 使用甲基化特异性的MLPA试剂盒(荷兰MRC-Holland公司,ME028),检测15q11.2-q13区域印记区缺陷、甲基化及单亲二倍体。
1.2.3 Sanger测序 应用Sanger测序对筛查到的变异进行验证,同时对其父母及外婆的相同基因位点进行验证。
1.2.4 文献复习 以“SLC6A1”、“癫痫”及“SLC6A1”、“epilepsy”为关键词,对万方数据库、中国知网和Pubmed建库至2023年1月收录的论文进行检索,并总结携带SLC6A1基因突变的国内患儿的临床表型和基因突变特点。
2 结 果
2.1 治疗及随访 患儿于2岁6月龄加用丙戊酸治疗(11 mg/kg·次,bid),随访至2022年10月,有20个月无发作,可独立行走、奔跑,语言障碍较前明显好转。
2.2 全外显子测序结果 发现先证者存在SLC6A1基因c. 889G>A(p. Gly297Arg)的杂合变异,属于常染色体显性遗传,此变异来自母亲及外婆(见图3)。已有文献报道过该变异位点,但Carvill等[5]报道的携带该变异位点的患儿有重度智力障碍,自闭症特征、攻击行为、脊柱侧弯、多种癫痫发作形式及对多种抗癫痫发作药物疗效差。Aguilera等[6]报道的患儿与本例相似,临床表型相对要轻,表现为Angelman样综合征,且丙戊酸单药控制良好。Piniella等[7]通过在过表达SLC6A1基因Gly297Arg突变体的人胚胎肾细胞293(human embryonic kidney 293,HEK293)中检测GABA摄取活性,发现与野生型(Wide-type,WT)相比,其摄取活性显著下降(低于WT的5%),证明该变异为致病性变异。

图3 家系全外显子组测序:先证者存在SLC6A1基因c. 889G>A(p. Gly297Arg)的杂合变异。此变异来自母亲及外婆
2.3 MLPA检测 本例患儿发作间期2次脑电图均表现为背景节律慢化(δ、θ混合慢波),醒睡期均可见双侧前头部、后头部或广泛性高波幅慢波、棘慢波,与Angelman综合征患儿脑电图表现相似,且患儿有全面性发育迟缓、言语障碍及刻板动作,但患儿并无Angelman综合征快乐面容。所以本例基因检测方法包括MLPA检测,排除Angelman综合征可能。结果染色体15q11-q13区域MLPA检测显示,在试剂盒检测范围内拷贝数和甲基化位点未见异常。
2.4 Sanger测序验证 先证者SLC6A1基因存在c. 889G>A(p. Gly297Arg)的杂合变异。先证者母亲及外婆均存在同样的杂合变异(见图3)。
2.5 文献复习 以“SLC6A1”“癫痫”以及“SLC6A1”“epilepsy”为关键词,对万方数据库、中国知网、Pubmed建库至2023年1月收录的论文进行检索,共检索到4篇[3,4,8,9]关于国内患儿携带SLC6A1基因变异的报告。结合本例患儿,目前国内共报道8例(男5例,女3例)SLC6A1突变的患儿(见表1)。8例患儿均有癫痫发作,起病年龄1~3岁。发作类型包括失神发作(5例,其中1例为不典型失神),肌阵挛发作(6例),肌阵挛-失张力发作(5例),全面强直阵挛发作(1例)。脑电图均可见广泛性棘慢波、多棘慢波发放,2例患者背景节律正常。8例患儿均存在发育迟缓,语言发育落后是突出特征。2例患儿报道存在自闭症样表现,1例患儿报道存在刻板样动作,1例患儿报道存在注意缺陷多动障碍。头部核磁检查无特异性异常表现。所有患儿均加用丙戊酸抗癫痫治疗,7例患儿对抗癫痫药反应良好,6例随访后期无发作,1例发作次数明显减少。1例患儿表现为难治性癫痫。除本例患儿SLC6A1基因存在家系遗传,其余患儿均为新发突变。1例为剪切变异,2例为无义变异,1例为缺失变异,其余4例均为错义变异。1例存在SLC6A1基因缺失变异的患儿还存在NOTCH1和PRIMPOL基因变异,所以该患者还具有主动脉瓣狭窄和高度近视的表现。
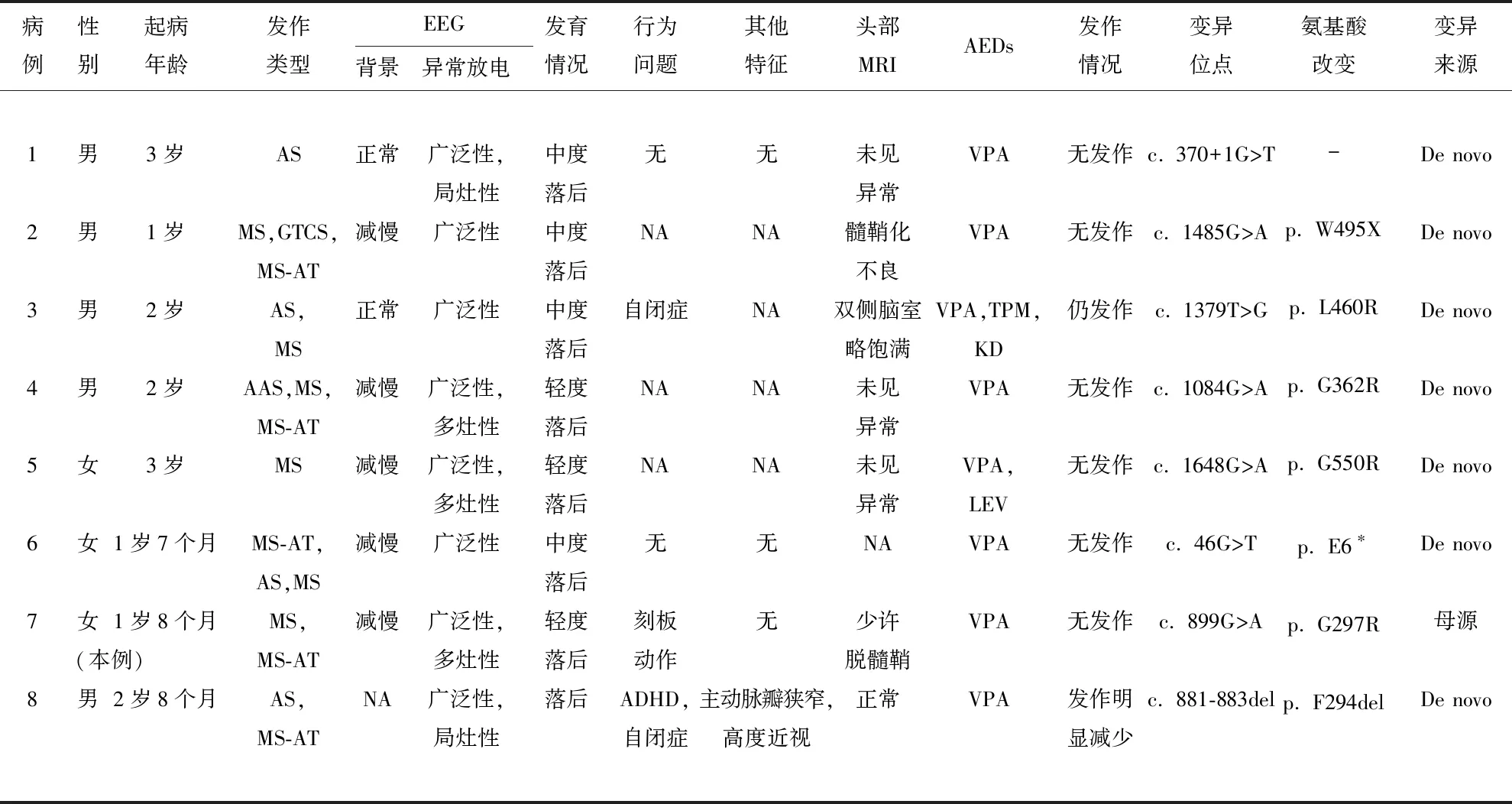
表1 8例SLC6A1基因变异的国内患儿临床特征及基因型
3 讨 论
SLC6A1相关疾病在2015年被首次描述[5]。SLC6A1 基因包含16个外显子,长度约46.5 kb,其编码的GABA 转运体 GAT-1 主要位于 GABA 能中间神经元的轴突和神经终端,GAT-1可从突触间隙重摄取GABA。GABA是哺乳动物中枢神经系统重要的抑制性神经递质,调节抑制性突触传递作用,从而防止脑内神经元过度兴奋[10]。目前研究表明,SLC6A1基因变异,可引起多种神经发育障碍。Goodspeed等总结了116例SLC6A1基因变异患者的临床资料,发现癫痫(91.1%)、发育迟缓及认知障碍(82.1%)、自闭症(22.8%)是最常见的临床特征[11]。
在一项队列研究中,采用高通量测序(next generation sequencing,NGS)联合拷贝数变异,对9 769例儿童癫痫患者进行筛查,发现SLC6A1被列为致病变异数较多的前10~20个基因之一[12]。
Johannesen等[13]报道16例SLC6A1基因突变患者,13例为点突变、3例为缺失突变,临床表现为癫痫2 例(发作形式包括肌阵挛发作、失神发作、非惊厥性癫痫持续状态、失张力发作、全面性强直阵挛发作、肌阵挛失张力发作),智力障碍16例(轻度智力障碍患者 11 例、中度智力障碍患者 4 例、1 例为严重的智力障碍),语言发育迟滞2 例,孤独症7例,注意力不集中2例等。但国内报道的8例患儿均有癫痫发作和发育迟缓,且语言发育迟滞是突出特点。
目前认为SLC6A1基因突变是发育性和癫痫性脑病的原因之一。据报道[11],癫痫首次发作的平均年龄是2.5岁。最常见的癫痫综合征是早发性癫痫伴肌阵挛失张力发作(20/82,24.3%),其次是遗传性全面性癫痫(19/82,23.1%)和非获得性局灶性癫痫(8/82,9.75%)。在56例获得详细癫痫发作形式的患者中,最常见的是不典型失神发作(71.7%)、失张力发作(44.4%)和肌阵挛发作(27.7%)。广泛性癫痫样放电(36/52,69.2%)是最常见的脑电图异常,可伴有背景节律变慢。这些特征与国内病例相似。但是对SLC6A1相关疾病而无癫痫发作的个体的脑电图特征目前知之甚少。本例患儿的脑电图特征是背景节律慢化,醒睡期可见双侧前头部、后头部或广泛性高波幅慢波、棘慢波,与Angelman综合征患儿脑电图表现相似,虽然患儿并无Angelman综合征的快乐面容,但考虑到患儿有全面性发育迟缓、言语障碍及刻板动作,所以本例基因检测方法包括MLPA检测,排除Angelman综合征可能。根据Aguilera等的报道[6],通过全外显子测序技术对14例Angelman样综合征患儿进行检测,发现了10种可引起Angelman样综合征的新的基因,其中之一就包括SLC6A1基因,且突变位点和本例患儿相同。Angelman综合征是母源UBE3A基因功能异常导致的神经发育遗传性疾病。Aguilera等认为,SLC6A1基因变异出现Angelman样综合征,可能由于SLC6A1基因受到UBE3A基因的调控。但文献中更多的关注基因型与表型之间的关系,对于脑电图特征并未进行描述。
在一项较大的关于自闭症测序的研究中(n=11 986),与23598个健康对照者相比,SLC6A1是自闭症患者变异较丰富的前10个基因之一[14]。有学者总结了27 例SLC6A1基因变异患儿的基因型与临床表型,18 例起病前有发育迟缓,11 例起病后出现发育倒退,9 例有自闭症表现,100%病例均遗留不同程度的智力障碍(intellectual disability,ID),语言障碍是最常见的特征[3]。本例患儿有明显语言障碍,虽有一些刻板动作,但自闭症相关量表评估未达到诊断标准。且国内病例仅有2例报道有自闭症样表现。
Goodspeed等[11]总结的116例SLC6A1基因变异患儿中发现85 种变异,变异类型包括88个错义变异,15个导致功能完全丧失的蛋白质截断变异,7个剪接位点的变异,3个拷贝数大的缺失变异,2个小的插入和缺失和1个同义变异,其中错义变异最常见(75.8%)。本例患儿为SLC6A1基因c.889G>A(p.Gly297Arg)变异为错义变异,既往已有文献报道[5~7],但虽为同一位点突变,既往报道的患儿临床表型严重程度与本例有差异,Carvill等[5]报道的患儿临床表现为重度智力障碍,自闭症特征、攻击行为、脊柱侧弯、多种癫痫发作形式及对多种抗发作药物疗效差。最近的一些研究已经证明不同类型的变异对GAT-1活性的影响程度不同,比如截断变异可能导致单倍剂量不足从而降低GAT-1表达,而一些错义突变可能通过显性-阴性效应导致GAT-1表达更低[15]。Piniella等[7]的研究结果显示,本例患儿携带的Gly297Arg变异对细胞内运输的影响是高度有害的,且该变异的GAT-1被保留在内质网中,完全无活性。这些研究表明,突变的GAT-1在内质网中的保留和降解会导致经过正确折叠、翻译、修饰的功能性GAT-1表达降低[16]。但这些仍不能解释即使是同一种变异,其临床表型也有差异,基因型和表型的相关性,还需进一步的深入研究。
目前还没有针对SLC6A1相关疾病的精准治疗药物,大多数患者需多学科联合治疗,包括神经病学、发育儿科、遗传学以及言语和职业治疗,以进行综合管理[17,18]。根据之前研究,丙戊酸是最有效的抗癫痫药物,拉莫三嗪和乙琥胺也显示效果良好[19],此外,其他调节GABA浓度的抗癫痫药,如氨己烯酸和噻加宾也可能发挥作用[20]。本例患儿采用丙戊酸单药治疗即有效,且患儿母亲及外婆仅幼时有癫痫发作,抗癫痫药治疗效果良好。但仍有许多SLC6A1基因变异患者表现为难治性癫痫。Mermer等[15,16,21]研究发现,用4-苯基丁酸(phenylbutyrate,PBA)处理携带致病性SLC6A1变异的细胞,可增加GAT-1的活性。这可能是由于PBA作为伴侣蛋白,减少了突变的GAT-1在内质网中的保留,并增加了功能性GAT-1的表达。这项研究可能会对药物治疗SLC6A1相关疾病做出进一步贡献。
综上所述,本研究中SLC6A1基因变异患儿主要表现为幼儿期起病的肌阵挛失张力癫痫,同时伴有轻度的认知障碍及语言障碍,药物治疗后无发作。患儿母亲及外婆临床表型较患儿轻,仅有幼时癫痫发作,而无明显认知障碍。通过文献复习,SLC6A1基因变异可导致多种神经发育障碍临床表型,可表现为癫痫(失神、失张力、肌阵挛及肌阵挛-失张力发作为主)、发育迟缓、认知障碍、孤独症或孤独症样表现及注意缺陷多动障碍等。SLC6A1基因突变可以为错义变异、无义变异、移码变异、剪切变异、染色体微缺失等。基因型与表型之间的关系还需进一步研究。化学伴侣蛋白PBA是治疗SLC6A1相关疾病的潜在有效药物。
猜你喜欢
心电与循环(2021年4期)2021-11-29
文萃报·周五版(2021年14期)2021-06-08
中国生殖健康(2020年7期)2021-01-18
数理医药学杂志(2019年12期)2019-12-05
中学数学杂志(2019年9期)2019-05-29
临床神经病学杂志(2017年6期)2018-01-13
海峡姐妹(2017年5期)2017-06-05
临床神经病学杂志(2017年2期)2017-04-27
丹青少年(2017年2期)2017-02-26
中国眼镜科技杂志(2016年17期)2016-10-24
