
湖泊沉积物磷形态分级提取及分析方法研究进展
2023-08-29 02:22方庆军唐亚东王小明徐一峰吴振斌
净水技术 2023年8期
方庆军,唐亚东,王小明,徐一峰,彭 来,吴振斌,张 义
(1.武汉理工大学资源与环境工程学院,湖北武汉 430070;2.中国科学院水生生物研究所淡水生态和生物技术国家重点实验室,湖北武汉 430072;3.华中农业大学资源与环境学院,湖北武汉 430070)
磷是水生环境中的必需营养元素,也是湖泊初级生产的限制性营养元素之一[1-2]。目前普遍观点认为,磷等营养元素过剩是湖泊富营养化问题产生的重要原因之一。一般认为,湖泊中磷元素的来源有外源输入和内源释放两个方面。外源部分主要是含磷的工业废水、农田灌溉污水、生活污水等输入,内源部分则来自于长期积累在沉积物中磷的释放。相关学者[3-4]通过对欧洲和北美35个湖泊进行长时间的检测发现,湖泊内源磷在外源输入得到有效控制之后的10~15年内仍会发生持续释放,在个别湖泊中可达到20年。沉积物内源磷在外部条件发生变化时,更容易通过间隙水交换进入上覆水中,提高了湖泊水生态系统初级生产力[5-6]。磷素在沉积物与和上覆水之间循环时,沉积物在“源”和“汇”角色之间转化。磷素在汇集到沉积物颗粒表面后,与发生吸附与解吸过程密切相关[7]。近年来出现的湖泊水体富营养化问题中,由外源输入引起的情况相对减少,更多则是沉积物内源磷持续释放引起较为严重的水华。因此,对沉积物磷内源释放的控制是解决水体持续性富营养化问题的关键。
研究沉积物磷的关键是准确进行沉积物磷的提取及测定。本文综述了磷分级提取方法的起源与发展,对磷连续提取方法做了较为详细介绍,分析提取方法优缺点,同时介绍了目前在分子水平上的磷形态主流分析方法,最后对未来的磷分级提取与分析方法进行了展望,以期为湖泊沉积物磷的变化规律研究提供一定的指导。
1 磷分级的生态学意义
在湖泊水环境中,磷的分布和赋存形态会直接或间接地影响到水体初级生产力及浮游生物的分布情况。湖泊通常被认为是污染源的“汇”,外源磷进入湖泊后会转变成不同形态。
不同赋存形态的磷,生物有效性和释放风险存在差异。例如,磷酸盐被吸附到沉积物表面形成的可交换态磷(exchangeable P,ex-P),可直接被浮游植物利用,在胶州湾的沉积物研究[8]显示,总磷(TP)中ex-P的占比会限制浮游植物的生长。铁结合态磷(Fe-P)在磷酸盐和铁氧化物或氢氧化物的共沉淀时形成,在还原条件下容易解吸,进入上覆水体[9]。钙结合态磷(Ca-P)则主要是生物磷灰石、碳酸钙配合磷酸盐或自生碳酸盐氟磷灰石等物质组成[10],其形成需要很长的时间,通常在深海沉积物中存在。
研究[11-12]表明,不同形态的磷生物活性及其在水环境迁移转化和循环路径不同,仅仅依靠TP含量来判断富营养化情况的传统方法已不能揭示水华暴发的过程与机制。因此,研究磷赋存形态,发展分级提取的方法有利于研究磷素行为,针对性解决水体富营养化问题,尤其对沉积物中磷内源释放的研究和治理具有重要意义。
2 沉积物磷的赋存形态
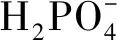
3 沉积物磷分级提取方法发展
一般认为,通过化学试剂提取不同形式沉积物磷的方法都起源于土壤中的磷分级提取方法[19],其基本原理是使用特定的化学试剂选择性地与沉积物中特定形态的磷结合,按照其结合的不同顺序进行连续提取,以获得沉积物中不同形态的磷。
沉积物磷的特征形态往往决定了磷的迁移、转化及归趋过程。仅通过TP或有效磷含量并不能全面、准确、长效地评估湖泊或者海洋沉积物的养分供应能力和生态环境风险,要深入研究沉积物磷对富营养化现象的作用机制,则需要能够科学表征、从分子层面准确认识磷形态[11]。因此,在研究初期出现了以提取分析为主的传统分析方法,随着技术发展,演变为现在以光谱法为主的分子层面分析技术。
3.1 IP分级提取方法发展
Dean[20]最早提出了土壤磷素分级,即用酸和碱试剂连续萃取来表征土壤中不同形式的IP,而由于当时磷的测定难度较高,直到1957年Chang提出的IP分级体系才得到广泛应用[20-21]。该分级体系将土壤中的IP分级提取分离为离散态磷形式,区分了Fe-P和Al-P,并首次明确提出了闭蓄态磷(O-P)的概念。这些磷形态主要包括铝结合态磷(Al-P)、Fe-P、Ca-P、还原性水溶态磷(RSP)和O-P。
1967年—1976年Williams等[22-24]对Chang-Jackson方法进行了改进,并应用到了沉积物中,将提取的磷分为磷灰石磷(AP)、非磷灰石磷(NAIP)和OP,并指出了在富含钙的沉积物中用NH4F提取铁铝结合态磷(Fe/Al-P)时易形成CaF2而重新吸附磷酸盐的缺点。
Hieltjes等[25]在1980年的研究则认为,当沉积物中的钙盐含量较高时,易出现NaOH提取的Fe-P被钙盐重新吸附,导致测定不准确的情况。其将Williams法改进,将沉积物磷分为不稳定性磷、Fe/Al-P、 Ca-P和残留态磷(residual-P)。
Psenner等[26]在1988年提出的磷提取方案与Chang关于在沉积物中磷形态的化学或矿物学定义的最初想法一致,即在第一步中使用1 mol/L NH4Cl代替H2O提取松散吸附的磷。该提取方案最初是针对硅质沉积物开发,此时碳酸盐对磷的再吸附在钙质沉积物中是不可避免的[27]。
在Hieltjes等[25]基础之上,通过用NaOH反复提取,Golterman[28]发现可以多提取出30%的正磷酸盐,表明用NaOH提取出的不仅是Fe-P。其认为在磷提取过程中应避免使用强酸和强碱溶液,而是通过使用螯合剂来提取Fe/Al-P。最初,Golterman[29]建议使用0.01 mol/L次氮基三乙酸钠溶液提取,随后改用0.05 mol/L次氮基三乙酸钙和0.05 mol/L次氮基三乙酸钠溶液依次溶解Fe/Ca-P,最终次氮基三乙酸及其盐又被乙二胺四乙酸(EDTA)所取代[28]。然而此法的缺点是EDTA会干扰正磷酸盐和Fe的测定。
1992年,Ruttenberg[30]为海洋沉积物开发了SEDEX提取法,单独量化了5种形态的磷,即不稳定性磷、Fe-P、碳酸氟磷灰岩磷(CFAP)、碎屑磷灰石磷(FAP)和OP。
欧洲委员会于1996年发起了沉积物磷的标准测试程序 (the standards, measurements and testing programme,SMT),以Williams法为基础确定了提取方案[31]。该方法简单实用,可操作性强,在分离不同的形态磷同时还能估计磷的潜在来源,因此,被广泛使用。SMT程序缺点在于使用强酸强碱作提取剂,对OP提取有影响且对OP的划分不够细致。表1总结了每种方法所涉及的步骤以及发展。

表1 IP分级提取方法发展
1998年,Jensen等[32]开发了一种用于海洋沉积物磷的6步连续提取方案,该方案是针对Psenner法的修改。主要修改是在第一步中使用1 mol/L MgCl2代替1 mol/L NH4Cl来提取松散吸附的磷,并在用NaOH提取之后和HCl提取之前加入额外的乙酸盐缓冲液[32]。此外,在每个提取步骤后用NaCl洗涤,可以减少磷酸盐的再吸附。
3.2 OP分级提取方法发展
1985年,Psenner等[33]提出了新的沉积物磷提取方法,他将磷分为水溶态磷(WSP)、RSP、Fe/Al-P、Ca-P和惰性磷。但该方法与Williams法类似,当沉积物碱质含量较高时,仍存在碳酸盐对磷的重吸附现象。
Golterman[29]在1977年—1988年的研究则认为,应该使用氨基三乙酸(NTA)替代强酸强碱作为提取剂,以减少对黏土结合态磷(一部分可交换态磷)和OP的破坏。然而该方法使用时样品准备程序复杂,提取时间较长,试验操作过程比较繁琐,可操作性不强。
1998年,Ivanoff等[34]重点研究了土壤中的OP连续提取方法,研究将沉积物磷形态按照活性强弱分为活性有机磷(LOP)、中活性有机磷(MLOP)和非活性有机磷(NOP),其中,MLOP为盐酸态有机磷和富里酸态有机磷,NOP中又细分为胡敏酸磷和残渣态磷。表2总结了OP分级提取方法的发展及优缺点。

表2 OP分级提取方法发展
4 沉积物磷的光谱学分析方法
尽管已经开发了许多连续分级方法来表征沉积物磷[35-36],但这些方法仅根据磷与基质组分间的相互作用强度,通过不同萃取剂中的溶解度差异来提取各种形态的磷,并不能确定特定的IP或OP的形态[35-37]。此外,在这些分级方法中,通常将未经消化的磷进行比色法测量并称为IP,TP和IP之间的差异称为OP。复杂的IP如焦磷酸盐和多聚磷酸盐等形态将包含在OP中,尽管它们不含碳[35]。此外,比色分析所需的低pH可能会降解OP或多聚磷酸盐,并释放正磷酸盐。湖泊中的OP可以被降解和矿化,最终水解或分解成为IP而被生物利用,但仍有相当一部分OP被保留在沉积物中,这部分OP被提取就较为困难。虽然可以通过分级提取或者计算的方式获得OP含量,但始终存在局限性[38-39]。识别特定磷形态的完整化学形式需要提取单个形式(如磷脂、RNA等)或先进的光谱技术,如X射线吸附光谱(X-ray absorption spectroscopy, XAS)或31P NMR光谱等[40-44]。以下将重点对几种应用广泛的典型磷形态表征技术进行介绍。
4.1 XAS
XAS可用于土壤、沉积物中多种元素的形态分析,因其需要较高的X射线通量和能量可调性,目前仅在同步加速器上可用。XAS的主要特征是“吸收边”,它基本上对应于光电子释放的阈值能量。XAS通常分为两个能量区域:X射线吸收近边结构光谱(X-ray absorption near edge structure, XANES)和扩展X射线吸收精细结构光谱(extended X-ray absorption fine structure, EXAFS)。XANES光谱部分包括边前区域、吸收边以及边后区域约为50 eV;EXAFS光谱为超出XANES的高能区域。
Ca-P、Al-P和Fe-P是沉积物中磷的主要赋存形式,具有不同的XANES特征峰,因此,可以通过吸收光谱来区分定量。对于Ca-P,XANES在白线峰位置的高能区域具有肩峰,磷灰石更加明显,而易溶态的磷酸钙则不明显。这些峰归因于碱土金属阳离子的激发光电子多次散射产生的连续共振[45-46]。边后的肩峰和2 160~2 170 eV其他次要光谱峰的位置和强度共同成为了Ca-P独特的指纹识别方法。Al-P在白线峰的低能区域产生肩峰,这在勃姆石(γ-AlOOH)吸附磷酸盐的XANES中不明显。此肩峰归因于磷 1 s电子激发为P(3p)-O(2p)-Al(3p)反键态[46-47]。XANES中磷酸铁和吸附在氧化铁上的磷酸盐的显著特征是在白线峰低能一侧2~5 eV处出现较弱的边前吸收峰。此边前峰的强度和能量位置与键合到金属磷酸盐中的过渡金属(如Zn、Co、Fe等)中的3 d电子数量有关。此边前峰归因于磷 1 s电子转变为Fe(3 d)型反键态,尽管根据电偶极子选择规则不允许s→d过渡,而p轨道混合成Fe(3 d)态允许这种过渡[48]。通过联用磷K边XANES和紫外可见吸收光谱并应用于一系列含有不同单齿和双齿键合比例的Fe-PO4络合物的水溶液,Khare等[47]认为这些特征涉及将1 s电子激发为Fe(4p)-O(2p)反键态。此外,对于具有不同结晶度、吸附相和络合物的矿物XANES,该特征峰的强度随Fe-O-P键数量的增加而增加。
除了对形态的鉴定,XAS还可以判断界面反应类型,如内圈、外圈配位、表面沉淀、共沉淀以及氧化还原等[49]。Khare等[50]采集了水铁矿吸附磷酸根样品的磷K边XANES,发现有磷的边前峰出现,表明磷酸根以内圈配位的形式吸附在水铁矿表面。且随着Fe/P的增加,XANES中白线峰的位置向高能一侧偏移了0.6 eV,远高于磷酸根质子化导致的白线峰偏移量(0.2 eV),同时结合理论计算,可以揭示磷酸根在水铁矿表面的配位方式由单齿单核配位向双齿双核配位转化[51]。
4.2 原子配对分布函数(pair distribution function, PDF)
PDF是基于X射线的总散射技术,是对粉晶X射线衍射结构模拟和XAS很好的补充,显示了与另一个原子在给定距离r处找到一个原子的概率[52-53]。最近,随着第三代同步加速器的发展,由于其能够提供更高通量和更短波长的X射线,以及可同时收集总散射图谱的快速读取探测器的应用,差分原子配对分布函数(differential pair distribution function, d-PDF)分析变得适用于更广泛的系统[54]。

4.3 核磁共振光谱(NMR)
NMR是研究原子核自旋行为以及原子核自旋与晶体结构关系的一种波谱学分析技术,其可以提供3种信息:化学位移、耦合常数以及积分曲线。其中化学位移对化学环境非常敏感,可以用来提供物质的分子结构信息,是NMR中最重要的参数[49]。
沉积物中相关磷化合物(有机或无机)的化学位移通常处于25~25 mg/L。这些磷包括:膦酸酯、正磷酸盐、正磷酸单酯、正磷酸二酯(磷脂和脱氧核糖核酸)、焦磷酸盐、多聚磷酸盐等。由于31P是唯一天然存在的磷同位素(100%自然丰度),理论上可以通过NMR法检测样品中的所有磷化合物。因此,如果选择了适当的采集参数,则每个峰面积与该特定类型的磷数量成正比,从而运用NMR可以定量鉴定样品中不同的磷形态。因此,31P NMR是研究土壤或沉积物磷的重要工具,极大地提高了对磷形态尤其是OP的认识。
31P NMR已被广泛用于表征土壤或沉积物中的OP组分[13,59-61]。其中,液相NMR主要针对OP测定,其应用最早是从生态学研究发展过来的。NMR具有检出限低、分辨率高等特点,对于水体和沉积物中的微量元素具有很好的分析效果。
化学位移与化学结构尤其是配位结构具有一定的对应性,可以用于判断物质的形态如内圈配位、外圈配位以及表面沉淀。Yan等[62]通过固态31P/27Al NMR研究了植酸在无定形氢氧化铝(AAH)表面的吸附,证实了在植酸在AAH表面由吸附态逐渐转化为类似于Al-IHP的表面沉淀。Lookman等[63]研究了吸附在无定型氧化铝表面的磷酸根形态,证明吸附方式以内圈配位为主。以上3种常见的光谱分析方法优缺点如表3所示。

表3 3种光谱学分析方法的优缺点
4.4 其他分析方法
除了上述介绍的几种分析方法外,一些表征方法如磷氧同位素以及理论计算如密度泛函理论等也越来越多用于磷形态的表征,尤其是磷形态的转化过程[64-65]。由于沉积物中磷酸盐响应成岩作用、溶解/沉淀和生物循环的不同反应性,磷酸盐的氧同位素比(δ18OP)可以携带这些过程的独特特征,揭示特定磷形态的起源[66-68]。Yuan等[69]利用磷酸盐的氧同位素以及沉积物化学表征如X-射线衍射和57Fe-Mössbauer光谱方法,分析了浅水湖泊沉积物的磷循环过程。结果表明,沉积物磷库的δ18OP值显着偏离平衡,因此,可以区分潜在的磷源或转化途径。自生磷的同位素值比平衡态低,说明有机质的再矿化和磷灰石的沉淀是自生磷形成的主要途径。18Al结合的磷(18.9‰~23.5‰)和Fe结合的磷(16.79‰~19.86‰)的OP值可能表明潜在的陆地来源,但后者更接近平衡值意味着来源于磷的还原溶解和释放。结果证实了好氧/缺氧振荡和溶解/再沉淀反应以及预期的同位素偏移。这些发现为更好地理解与浅淡水湖泊富营养化相关磷的起源和生物地球化学循环具有重要意义。
5 展望
总体而言,国内外对于沉积物磷的赋存形态、磷素提取及分析方法的发展始于二十世纪六七十年代,到目前为止,已经趋于成熟。总体来看,我国在沉积物磷的赋存形态及转化的机制方面的研究水平和深度仍然有待提高,应该向着分子水平继续深入,细化对各种形态磷的分类,创新不同形态磷的分析方法,深刻阐释湖泊沉积物磷素转化机制,这对控制和解决湖泊富营养化问题具有深远的意义。
猜你喜欢
海洋通报(2022年2期)2022-06-30
海洋石油(2021年3期)2021-11-05
河北环境工程学院学报(2021年1期)2021-03-19
少儿美术(快乐历史地理)(2019年4期)2019-08-27
疯狂英语·新读写(2018年3期)2018-11-29
阅读(低年级)(2018年4期)2018-05-14
小学阅读指南·低年级版(2017年2期)2017-03-23
电镀与环保(2016年2期)2017-01-20
环境科技(2015年4期)2015-11-08
无机化学学报(2014年12期)2014-02-28
