
地下水系统中富里酸-铁-砷的共沉淀行为
2024-02-24 14:19赵华淼甘义群
安全与环境工程 2024年1期
赵华淼,甘义群,赵 琪
(中国地质大学(武汉)环境学院,湖北 武汉 430078)
砷(As)是地下水系统中常见的一种有毒污染物,长期饮用高砷地下水会严重损害人的身体健康[1-2]。铁(氢)氧化物和有机质是土壤与沉积物中的反应性组分[3-4],与砷的地球化学行为密切相关[5-6]。铁(氢)氧化物对砷的固定至关重要[7],当含水层氧化还原条件发生波动时,地下水中的As(Ⅲ)易氧化为As(Ⅴ),Fe(Ⅱ)氧化为Fe(Ⅲ),随后As(Ⅴ)与Fe(Ⅲ)的共沉淀可有效减少砷向水生系统的释放[8];而有机质易与砷络合,增加其迁移率[9]。然而,铁(氢)氧化物与有机质通常不单独存在于自然环境中,两者多通过吸附或共沉淀反应相结合[10-11],并会影响砷的环境行为[12]。
近些年,许多学者针对有机质、铁(氢)氧化物和砷的相互作用机制进行了大量的试验研究[13-14],结果表明:有机质能够通过静电作用、配位交换等方式直接与砷竞争铁(氢)氧化物表面吸附位点[14-15];有机质还可以改变铁(氢)氧化物的结构与性质[16],使其结晶度降低,表面负电荷增多,从而减少对砷的吸附[12,17];此外,若考虑环境的氧化还原条件,有机质可作为电子供体或电子穿梭体促进铁(氢)氧化物还原溶解与砷释放[18-19]。由此可见,已有研究多集中于砷吸附或溶解过程,且试验变量主要为有机质种类与浓度,但对砷的共沉淀过程,尤其是在不同Fe(Ⅲ)浓度和pH值条件下C-Fe-As耦合机制的研究十分薄弱,而这是准确理解与预测砷迁移转化规律的基础。
针对以上问题,本文将地下水系统中有机质-铁(氢)氧化物-砷三元耦合反应体系作为研究对象,选取高砷含水层中常见的富里酸(FA)作为代表性有机质进行室内控制试验[15],探究Fe(Ⅲ)浓度与pH值对FA-Fe(Ⅲ)-As(Ⅴ)共沉淀行为的影响,并通过分析共沉淀反应后有机质的组分变化以及有机质-铁(氢)氧化物共沉淀(organo-Fe hydroxides coprecipitates,简称OFC)的结构与性质,阐明共沉淀过程中砷与有机质的相互作用机制,从有机质组分分馏的新视角揭示OFC的固砷机理。研究结果对理解地下水系统中砷与碳元素的地球化学循环规律具有重要意义,并可为地下水环境中砷污染的治理与防治提供相关理论基础。
1 材料与方法
1.1 试验材料
准确称取0.1 g富里酸(FA)标准固体(阿拉丁,CAS:1415-93-6)溶于100 mL 0.1 mol/L的NaOH溶液中,后置于恒温振荡箱中避光振荡(200 r/min)24 h,使其充分溶解,后用0.45 μm水系纤维滤膜过滤至棕色玻璃瓶中,得到1 g/L的FA储备液,于4 ℃下保存,用前将FA储备液稀释至15 mg C/L待用。为了防止存放时间过长导致有机质分解,FA储备液均应在5 d内使用完毕,并保证每一批次试验的FA溶液源自同一储备液。使用Fe(NO3)3·9H2O、Na3AsO4·12H2O固体配制0.2 mol/L Fe(Ⅲ)、1 g/L As(Ⅴ)储备液,调节Fe(Ⅲ)储备液的pH值小于2.0,以避免Fe(Ⅲ)发生水解沉淀。
1.2 不同Fe(Ⅲ)浓度条件下的共沉淀试验
根据自然环境中常见的C/Fe范围(0.2~25.0)[11,20],在固定初始FA浓度的条件下,于0~1.6 mmol/L范围内设置16种初始Fe(Ⅲ)浓度进行共沉淀试验。试验具体操作步骤如下:取25 mL FA溶液置于一系列50 mL离心管中,同时加入不同体积Fe(Ⅲ)储备液与等体积As(Ⅴ)储备液,初始As(Ⅴ)浓度为2.5 mg/L,为了减少FA的稀释效应,加入的Fe(Ⅲ)与As(Ⅴ)溶液体积均控制在200 μL以内,背景溶液为0.01 mol/L的NaCl溶液;3种溶液混合均匀后逐滴加入0.1 mol/L NaOH或HCl调节pH值至5.0,在该pH值条件下Fe(Ⅲ)和FA共沉淀显著发生[10];待pH值稳定后,将离心管放置在25 ℃恒温振荡箱中避光振荡24 h,转速设为200 r/min,振荡结束后以4 000 r/min转速离心15 min,取上清液过0.45 μm水系纤维滤膜,用于水化学分析,沉淀物冷冻干燥并研磨至200目,用于固相表征分析。试验设置不含As(Ⅴ)和不含FA的反应体系为对照组。
1.3 不同pH值条件下的共沉淀试验
在pH值为5.0、7.0、9.0时,研究不同pH值对共沉淀反应体系中As(Ⅴ)与FA相互作用的影响。在相同pH值条件下,体系初始Fe(Ⅲ)浓度分别设为0.08、0.12、0.16、0.24、0.32、0.63、1.25 mmol/L,初始As(Ⅴ)浓度为2.5 mg/L,振荡前调节pH值为所需值,其余步骤与第1.2节中一致。
1.4 样品表征方法
样品X射线衍射(XRD)图谱使用Bruker D8 Advance仪测定,并使用HighScore软件对样品XRD图谱进行分析;扫描电子显微镜与能谱分析(SEM-EDS)使用TESCAN MIRA LMS扫描电镜仪拍摄沉淀物样品形貌与能谱;样品X射线光电子能谱(XPS)使用Thermo Scientific K-Alpha仪测定,并将采集到的能谱使用Bruker D8 Avantage仪进行分析;样品中溶解性有机碳(DOC)浓度使用Elementar Vario TOC分析仪测定;样品中As(Ⅴ)浓度使用北京吉天AFS-9700原子荧光光度计测定;样品中Fe(Ⅲ)浓度采用邻菲罗啉分光光度法测定,使用仪器为紫外可见分光光度计(UV-1800);样品EEMs光谱使用日立F-4600荧光光度计测定,荧光数据采用DOM Fluor工具箱进行平行因子分析,单个荧光组分的相对浓度用最大荧光强度(Fmax)表示。
2 结果与讨论
2.1 不同Fe(Ⅲ)浓度条件下FA-Fe(Ⅲ)-As(Ⅴ)的共沉淀行为
2.1.1 Fe(Ⅲ)浓度对As和FA固定量的影响
图1为不同Fe(Ⅲ)浓度条件下As和FA固定量的变化曲线图。
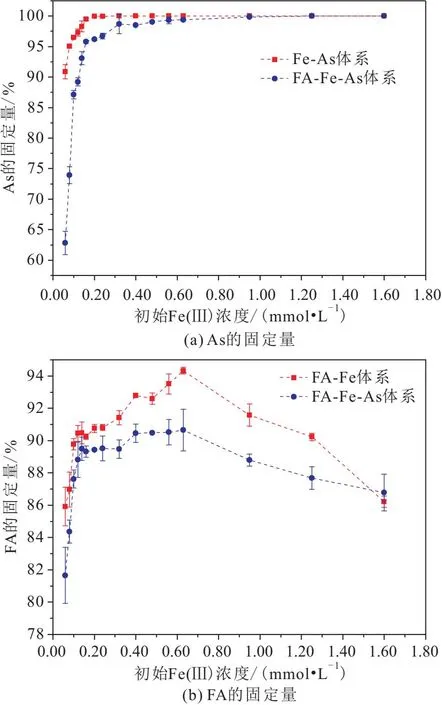
图1 不同Fe(Ⅲ)浓度条件下As和FA固定量的变化曲线Fig.1 Change curves of As and FA sequestration propor-tions under different Fe(Ⅲ) concentrations
由图1可以看出:
1) 共沉淀后砷和有机质FA的固存规律很大程度上受初始Fe(Ⅲ)浓度的影响。在Fe-As体系中,当初始Fe(Ⅲ)浓度从0.06 mmol/L增至0.24 mmol/L时,As(Ⅴ)的固定量从90.88%增至100%,说明Fe-As体系有较强的固砷能力,这可能是由于在酸性环境中,Fe(Ⅲ)与As(Ⅴ)主要以砷酸铁相共沉淀,除了部分As(Ⅴ)吸附在水铁矿表面外,大量的As(Ⅴ)能够固定在水铁矿的晶格中[21];FA参与共沉淀后,随着初始Fe(Ⅲ)浓度从0.06 mmol/L增至1.25 mmol/L,As(Ⅴ)的固定量从62.82%增至100%,与Fe-As体系相比,FA-Fe-As体系中As(Ⅴ)的固定量达到100%需要更多Fe(Ⅲ)输入,意味着FA的存在抑制了As(Ⅴ)与Fe(Ⅲ)的共沉淀,且抑制作用在Fe(Ⅲ)浓度低时更显著,当Fe(Ⅲ)浓度超过1.25 mmol/L后抑制作用可忽略不计。
2) 有机质FA的固存规律不同于砷。对于FA-Fe体系,初始Fe(Ⅲ)浓度从0.06 mmol/L增至1.60 mmol/L的过程中,FA的固定量为85.92%~94.33%,总体呈先增大后减小的变化规律,减小趋势在初始Fe(Ⅲ)浓度大于0.63 mmol/L后出现,这是由于共沉淀过程中Fe(Ⅲ)浓度较低时,容易形成Fe(Ⅲ)-FA络合物,较高含量的络合物抑制FA与Fe(Ⅲ)发生水解共沉淀[22],OFC的形成较少,随着Fe(Ⅲ)浓度的升高,水解共沉淀作用增强;当初始Fe(Ⅲ)浓度增至0.63 mmol/L后,FA与Fe(Ⅲ)的相互作用方式会再次改变,由水解共沉淀逐渐向吸附转化[23],而吸附作用对有机质的固存效率显著低于共沉淀作用。As(Ⅴ)参与共沉淀后,FA的固定量在81.65%~90.65%范围内变化,整体明显减小,说明As(Ⅴ)和FA在与水铁矿结合的过程中存在竞争关系,但在初始Fe(Ⅲ)浓度为1.60 mmol/L的情况下竞争关系不明显,可能由于此时水铁矿含量较高,无论其表面还是内部均有充足的吸附位点。
2.1.2 Fe(Ⅲ)浓度对FA组分分馏的影响
本试验使用三维荧光光谱对共沉淀反应后溶液中有机质(FA)的成分进行表征,并采用平行因子分析法,从EEM数据集中提取4种荧光组分,图2为C1~C4 4种荧光组分的三维荧光光谱图及对应的发射、激发特征图,表1为FA的4种荧光组分的特征描述。
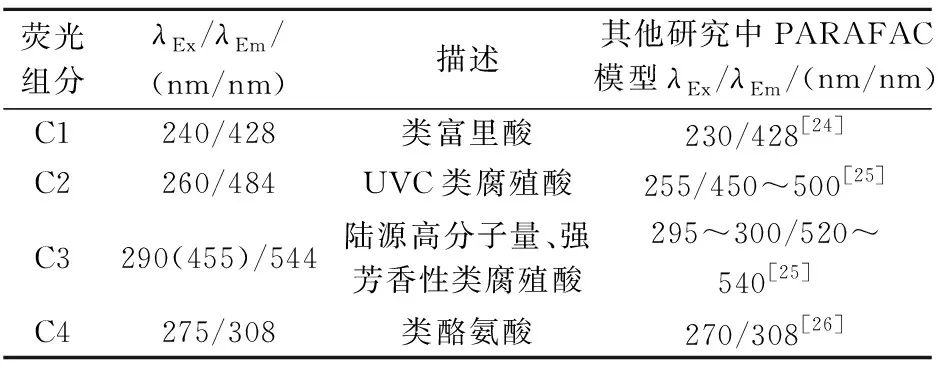
表1 FA的4种荧光组分的特征描述
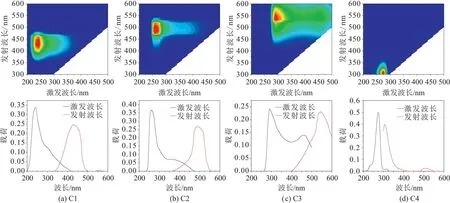
图2 FA三维荧光-平行因子分析法提取的4种FA荧光组分及对应的发射/激发载荷Fig.2 Four fluorescent components and the corresponding emission/excitation loadings of FA extracted by three-dimensional fluorescence parallel factor analysis
不同Fe(Ⅲ)浓度条件下FA各组分荧光强度的变化如图3所示。
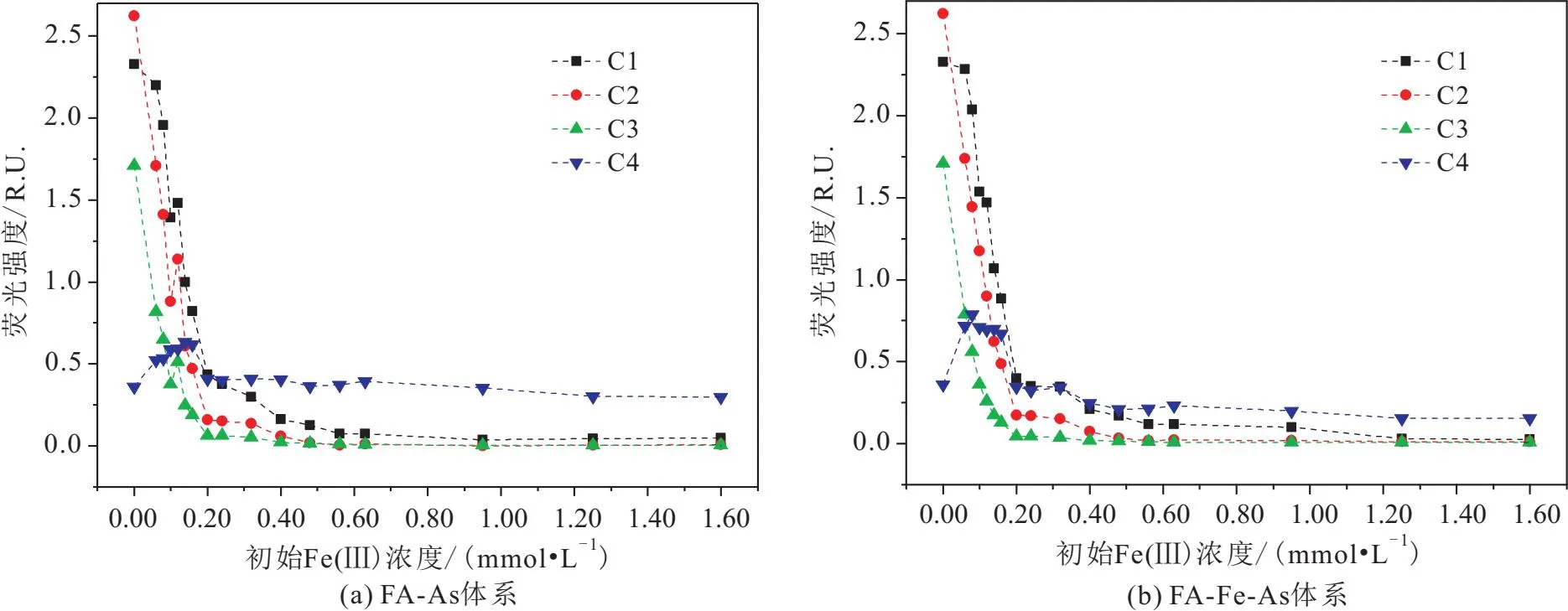
图3 不同Fe(Ⅲ)浓度条件下FA各组分荧光强度的变化曲线Fig.3 Change curves of fluorescence intensity of FA components under different Fe(Ⅲ) concentrations
由图3可知:FA荧光组分的荧光强度受初始Fe(Ⅲ)浓度的影响显著。在FA-Fe体系中:随着初始Fe(Ⅲ)浓度的增大,溶液中C1~C3组分荧光强度逐渐减弱后趋于平稳,C3组分荧光强度的减弱程度最高,初始Fe(Ⅲ)浓度超过0.20 mmol/L后溶液的总荧光强度明显降低;原FA溶液中检测出C4组分荧光强度最低,但在Fe(Ⅲ)加入后溶液中C4组分荧光强度上升,造成该现象的原因可能是本研究使用的FA以类腐殖酸与类富里酸为主要成分,原FA溶液中这些组分的荧光强度高,遮盖了类蛋白质组分的荧光峰[27],共沉淀反应后类腐殖质组分被从溶液中去除,尤其是类腐殖酸组分荧光强度明显降低,因此类蛋白质组分的荧光峰得以显现;初始Fe(Ⅲ)浓度超过0.20 mmol/L后溶液中C4组分荧光强度开始缓慢减弱,此时其余组分已被水铁矿大量固定。在FA-Fe-As体系中:相同初始Fe(Ⅲ)浓度的条件下,溶液中C2、C3组分荧光强度差异不明显,但C1组分荧光强度普遍高于FA-Fe体系,C4组分则相反。
以上结果表明:与Fe(Ⅲ)共沉淀后FA会发生显著的组分分馏,陆源高分子量类腐殖酸组分会优先参与共沉淀,其次是UVC类腐殖酸与类富里酸组分,最后是类蛋白质组分。在初始Fe(Ⅲ)浓度小于0.20 mmol/L时,Fe(Ⅲ)-FA络合物在沉淀物中仍占有一定比例,而络合反应导致有机质组分分馏程度低于水解共沉淀,所以共沉淀反应后溶液中FA各组分荧光强度仍处于较高水平。As(Ⅴ)的存在并未改变组分分馏模式,但抑制了类富里酸与类腐殖酸组分的共沉淀,并对类富里酸组分的抑制能力更强。因此,在共沉淀反应中As(Ⅴ)主要通过与类富里酸、类腐殖酸组分竞争与水铁矿的结合位点,减少OFC中这两种组分的固存而被固定。
2.1.3 Fe(Ⅲ)浓度对共沉淀产物基本性质的影响
图4为共沉淀产物、FA和水铁矿(Fh)的XRD图谱。共沉淀产物的XRD分析结果表明,初始Fe(Ⅲ)浓度影响了共沉淀产物的结晶度。当Fe(Ⅲ)浓度为0.08 mmol/L(该浓度条件下,含As的共沉淀产物为OFC-As-1,不含As的共沉淀产物为OFC-1)时,图谱中未出现显著的矿物衍射峰,仅存在一个宽化衍射峰,其晶体结构与FA固体粉末类似,只是宽化衍射峰的强度更低,说明该沉淀物结晶度很低,具有“类有机质”结构。当Fe(Ⅲ)浓度为0.32 mmol/L(该浓度条件下,含As的共沉淀产物为OFC-As-2,不含As的共沉淀产物为OFC-2)、1.25 mmol/L(该浓度条件下,含As的共沉淀产物为OFC-As-3,不含As的共沉淀产物为OFC-3)时,图谱在2θ为35°和62°处有比较明显的宽峰,说明沉淀物中存在二线水铁矿,具有“类矿物”结构,但两个特征峰的强度均低于水铁矿,前者特征峰的强度更低,意味着FA的存在抑制了矿物组分向结晶态铁(氢)氧化物的转化[28],FA的相对浓度升高导致沉淀物中水铁矿的比例降低。对比含As(Ⅴ)与不含As(Ⅴ)的OFC的XRD图谱发现,As(Ⅴ)的存在增加了矿物的结晶度,Fe(Ⅲ)浓度较低时,As(Ⅴ)对矿物结晶度的影响更显著,该现象可能是由于As(Ⅴ)抑制了部分FA与Fe(Ⅲ)的共沉淀所导致的。
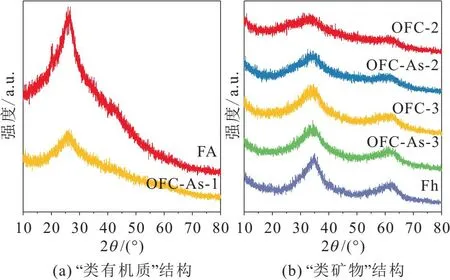
图4 共沉淀产物、FA和水铁矿的XRD图谱Fig.4 XRD patterns of OFC,FA and ferrihydrite
图5为共沉淀产物的SEM图像和EDS图谱。

注:黄色的“+”为EDS选定点。图5 共沉淀产物的SEM图像与EDS图谱Fig.5 SEM images and EDS spectra of the OFC
由图5可见:初始Fe(Ⅲ)浓度改变了共沉淀产物的形貌特征。共沉淀产物均由纳米级无规则形状颗粒组成,这些颗粒倾向于聚集在一起形成较大聚集体,不同的是,C元素相对含量增大后,矿物的表面孔隙或沟壑中会进入一些片状或块状的FA颗粒,使矿物表面的粗糙程度增大,聚集体的形成减少,结晶程度降低,说明共存的FA可能抑制铁(氢)氧化物的聚集与矿化;EDS面分析图谱显示,沉淀物表面分布的元素包括C、O、Fe、As,Fe(Ⅲ)浓度较低的情况下,C与As元素在矿物表面的质量百分数更大,分布更密集,Fe则相反。
在每种共沉淀产物表面分别选取2~3个点位进行EDS点分析,其结果如表2所示。
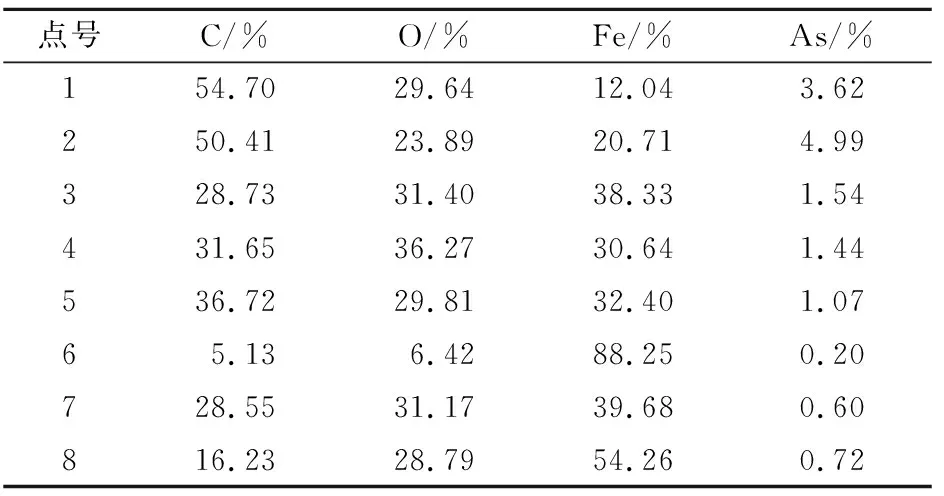
表2 EDS选定点各元素质量百分数
由表2可知,各元素在6~8号点的质量百分数变化程度较大,但在1~2、3~5号点仅有小幅波动。
以OFC-2与OFC-As-2为例,进一步使用XPS图谱来表征共沉淀产物表面元素组成与化学键的变化情况,其结果见图6。
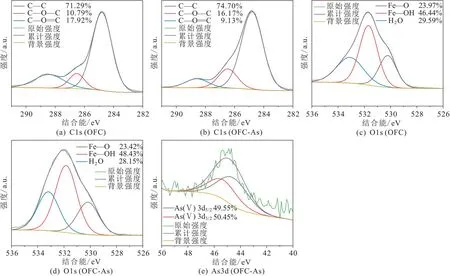
图6 共沉淀产物C1s、O1s、As3d的XPS图谱Fig.6 C1s,O1s,As3d XPS spectra of the OFC
由图6可见:C1s能谱分为284.8 (284.8) eV、286.6 (286.5) eV和288.4 (288.6) eV 3个峰,代表C—C、C—O—C、O—C=O的特征峰,其中C—C键在共沉淀产物表面含量最高,而含氧官能团中O—C=O键的含量高于C—O—C键,造成该现象的原因可能是FA溶液中羧基碳含量本身较高,同时Fe(Ⅲ)会优先共沉淀羧基碳[22],而As(Ⅴ)参与共沉淀反应后,O—C=O键含量减小,C—C键与C—O—C键含量增大,说明As(Ⅴ)抑制了部分FA溶液中的羧基碳与Fe(Ⅲ)共沉淀;O1s能谱分为530.2 (530.2) eV、531.7 (531.8) eV和533.1 (533.2) eV 3个峰,代表Fe—O、Fe—OH、H2O的特征峰,Fe—OH键的含量最高,而As(Ⅴ)参与共沉淀反应后,Fe—OH键含量增大,Fe—O键含量减小,已有研究表明Fe—O—H键与Fe—O—As键的结合能接近,无法区分它们的相对贡献率[17],推测沉淀物表面的As可能形成Fe—O—As络合物被固定;As3d能谱分为44.8 eV和45.6 eV 2个峰,且As(Ⅴ)在共沉淀反应后价态未发生变化。
1.3 统计学处理 采用SPSS22.0统计软件进行分析。以描述性与推论性统计的百分比、平均值、标准偏差、t检验、皮尔森积差相关分析与单因子变异数分析来呈现医护人员在相关因素的分布情形、个人基本资料与其认知及3个相关因素结构面间的相关性。
2.2 不同pH值条件下FA-Fe(Ⅲ)-As(Ⅴ)的共沉淀行为
2.2.1 pH值对As与FA固定量的影响
图7为不同pH值条件下共沉淀反应体系中As、FA与Fe含量的变化曲线图。
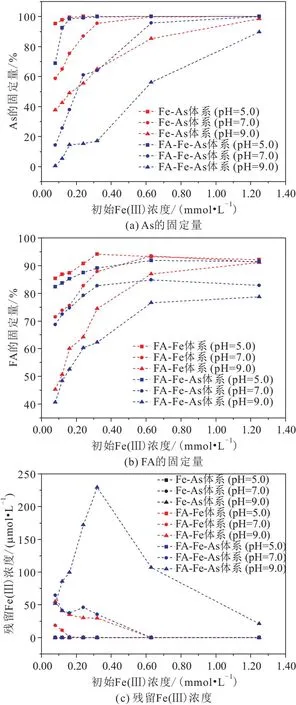
图7 不同pH值条件下共沉淀反应体系中As、FA与Fe含量的变化曲线Fig.7 Change curves of As,FA and Fe content in the coprecipitation system under different pH
由图7可以看出:
1) 反应体系初始pH值显著影响共沉淀反应的固砷效率。对于Fe-As体系,不同pH值条件下As(Ⅴ)的固定量均随初始Fe(Ⅲ)浓度的增大而增大,且溶液中几乎没有Fe(Ⅲ)残留;但较高的pH值不利于As(Ⅴ)的固定,初始Fe(Ⅲ)浓度较低时,pH值的影响更明显,可能是pH值改变As(Ⅴ)与Fe(Ⅲ)的相互作用方式导致[21]。与酸性环境不同,在中性与碱性环境中,Fe(Ⅲ)先沉淀形成水铁矿,As(Ⅴ)主要通过吸附作用固定在水铁矿表面,碱性环境中水铁矿吸附As(Ⅴ)的能力较弱[29]。FA参与共沉淀反应后,3种pH值条件下FA对As(Ⅴ)的固定产生不同程度的抑制作用,在中性与碱性环境中抑制作用更显著,当pH值为7.0时溶液中有35.19~64.71 μmol/L Fe(Ⅲ)残留,当pH值为9.0时溶液中有20.90~229.32 μmol/L Fe(Ⅲ)残留,表明FA阻碍了Fe(Ⅲ)的沉淀。
2) 与As(Ⅴ)变化规律相似,反应体系初始pH值也影响有机质的固存,对比两种反应体系发现,与酸性环境相反,在中性与碱性环境中,初始Fe(Ⅲ)浓度较高时,As(Ⅴ)对有机质的固存影响较大,这是因为在C/Fe值较低的情况下,FA与Fe(Ⅲ)的相互作用方式由水解共沉淀转变为吸附[23],更多的FA存在于水铁矿表面,此时As(Ⅴ)的固定方式主要为表面吸附,所以OFC通过减少表面吸附的FA来固定As(Ⅴ)。
2.2.2 pH值对FA组分分馏的影响
不同pH值条件下共沉淀反应体系中FA各组分荧光强度与相对浓度的变化,见图8。
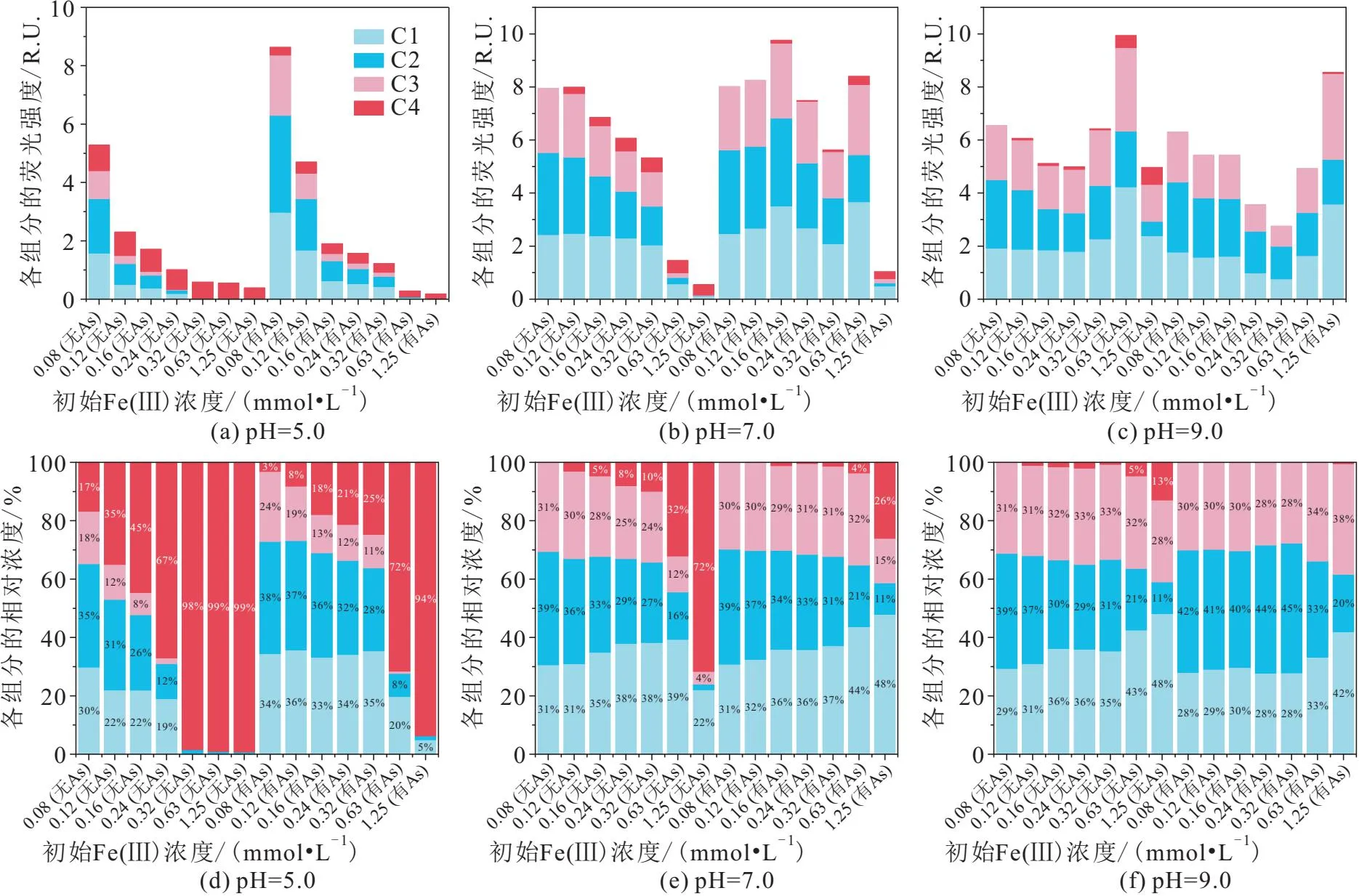
图8 不同pH值条件下共沉淀反应体系中FA各组分荧光强度与相对浓度的变化Fig.8 Changes of fluorescence intensity and relative abundance of FA components in the coprecipitation system under different pH values
1) 在不同pH值条件下共沉淀反应体系中,FA各荧光组分均发生不同程度的分馏。在高浓度Fe(Ⅲ)输入的酸性环境中,FA的分馏程度更高,陆源高分子量、强芳香性组分优先共沉淀;碱性环境中C1~C3组分的相对浓度明显高于酸性环境,C4组分则相反,这主要是因为在碱性环境中Fe(Ⅲ)溶解度较大,与FA更容易发生络合反应[30],Fe(Ⅲ)与陆源高分子量、强芳香性组分的络合减弱了FA的分馏,Fe-FA二元络合物的形成增加了溶液中有机质的含量,从而导致C1~C3组分的相对浓度升高。
2) pH值对有机质结构与性质的影响也不可忽视,即反应体系pH值升高,有机质的苯环结构增多,芳香性与共轭度增强[31]。
3) As(Ⅴ)的存在未改变FA的分馏模式,但影响了各组分的荧光强度与相对浓度,其在不同pH值条件下的影响程度也不同。As(Ⅴ)参与共沉淀后,在低浓度Fe(Ⅲ)输入的酸性环境中,C1~C3组分的荧光强度与相对浓度均升高,说明As(Ⅴ)使OFC内部结合的C1~C3组分解吸,初始Fe(Ⅲ)浓度达到0.63 mmol/L后,As(Ⅴ)的共沉淀对C3组分的相对浓度影响不大,此时OFC主要通过减少C1与C2组分的固存来固定As(Ⅴ);在中性环境中,C1~C3组分的荧光强度均增高,初始Fe(Ⅲ)浓度较低时,As(Ⅴ)的加入抑制Fe(Ⅲ)的沉淀,可能促进了As-Fe-FA三元络合物的形成,因此C1~C3组分的共沉淀减少,初始Fe(Ⅲ)浓度达到0.63 mmol/L后,溶液中的Fe(Ⅲ)完全沉淀,As(Ⅴ)主要与C1~C3组分竞争水铁矿的结合位点而被固定;在碱性环境中,C1组分的荧光强度与相对浓度均降低,C2、C3组分的荧光强度变化规律不明显,但相对浓度升高。3种环境中C4组分的变化规律单一,As(Ⅴ)参与共沉淀后其荧光强度与相对浓度均降低,推测可能是As(Ⅴ)与类蛋白质组分络合后,对该组分产生明显的荧光猝灭效应[31],尤其在碱性条件下溶液中仍有较高浓度的As(Ⅴ)残留。
2.3 FA-Fe(Ⅲ)-As(Ⅴ)共沉淀机理分析
FA-Fe(Ⅲ)-As(Ⅴ)共沉淀机理如图9所示。初始Fe(Ⅲ)浓度较低时,FA与Fe(Ⅲ)主要发生络合反应;初始Fe(Ⅲ)浓度增至0.63 mmol/L的过程中,FA与Fe(Ⅲ)主要发生水解共沉淀反应,高分子量、强芳香性组分的羧基或酚羟基通过配体交换与水铁矿结合形成OFC;初始Fe(Ⅲ)浓度超过0.63 mmol/L后,主要发生吸附反应。
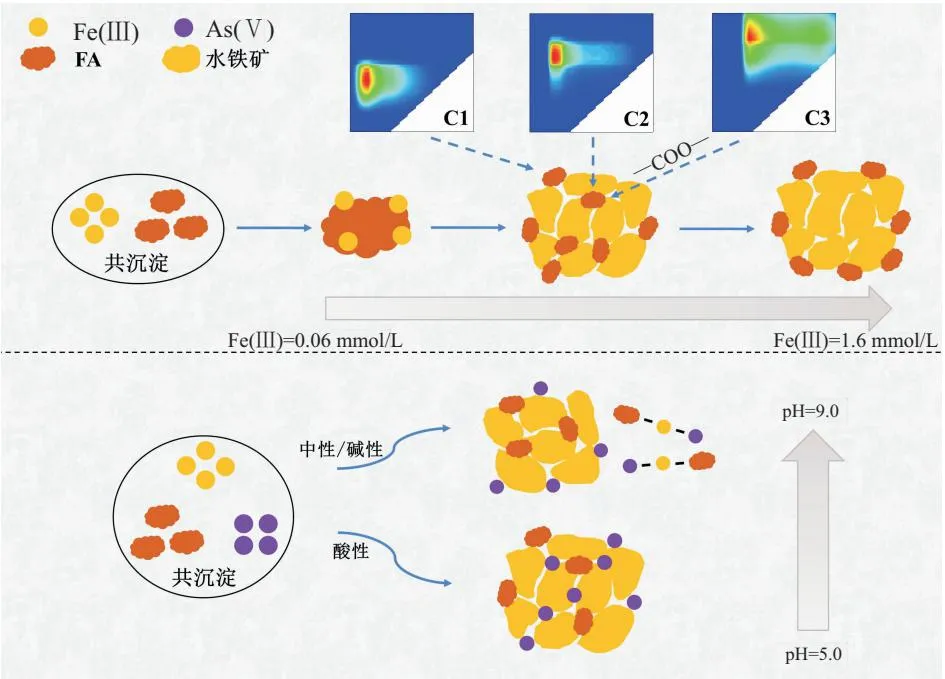
图9 FA-Fe(Ⅲ)-As(Ⅴ)共沉淀机理示意图Fig.9 Schematic diagram of FA-Fe(Ⅲ)-As(Ⅴ) coprecipitation mechanism
OFC固定砷时,砷在酸性环境中可分布于OFC的内部或表面,与FA的存在方式类似;但在中性与碱性环境中,砷以表面分布为主,FA促进OFC中的水铁矿配位溶解,归因于FA中的羧基在pH值较高时易电离H+,水铁矿失去OH-,两者配位形成FA-Fe(Ⅲ)络合物,随后在FA亲水基团的作用下络合物脱离OFC以溶解的形式存在于溶液中,Fe(Ⅲ)充当连接FA与As(Ⅴ)的阳离子桥,As-Fe-FA络合物的形成减少了OFC对砷的固定,但固砷过程会导致OFC中的FA解吸,因此砷通过占据FA与水铁矿的结合位点而被OFC固定。
3 结 论
本文以地下水系统中C-Fe-As耦合反应体系作为研究对象,围绕共沉淀反应进行批试验,探究了不同Fe(Ⅲ)浓度和pH值条件下FA-Fe(Ⅲ)-As(Ⅴ)的共沉淀行为,并结合3D-EEMs、XRD、SEM-EDS和XPS表征手段,从有机质组分分馏的视角揭示了OFC的固砷机理,主要取得了以下认识:
1) FA-Fe(Ⅲ)-As(Ⅴ)的共沉淀行为受初始Fe(Ⅲ)浓度的影响明显。随着初始Fe(Ⅲ)浓度的增大,As(Ⅴ)的固定量逐渐增大至100%,FA的组分分馏程度增大,且富含羧基的类腐殖质组分优先共沉淀;OFC呈现“类有机质”向“类矿物”结构的转变,水铁矿的比例升高,其表面的As(Ⅴ)分布更加松散。
2) pH值会进一步影响FA-Fe(Ⅲ)-As(Ⅴ)的共沉淀行为。较高的pH值不利于砷的固定以及FA的组分分馏。在中性与碱性环境中,FA减少了Fe(Ⅲ)的沉淀,促进了可溶性As-Fe-FA络合物的形成。
3) 在共沉淀过程中,As(Ⅴ)与FA存在竞争关系,As(Ⅴ)抑制FA中类腐殖质组分的共沉淀,并对类富里酸组分的抑制能力更强;As(Ⅴ)主要通过与类腐殖质组分竞争OFC中水铁矿的结合位点,减少该组分的固存而被固定。
猜你喜欢
特产研究(2024年1期)2024-03-12
煤气与热力(2021年12期)2022-01-19
成都大学学报(自然科学版)(2021年1期)2021-05-22
读者·校园版(2020年21期)2020-10-29
天然产物研究与开发(2019年10期)2019-11-05
中成药(2018年8期)2018-08-29
中成药(2018年2期)2018-05-09
河北地质(2016年3期)2016-04-23
有色金属设计(2014年4期)2014-03-11
金属矿山(2013年6期)2013-03-11
