
2-甲酰基-1,1′-联萘手性醛催化剂的合成
2024-02-28 01:40王煜洋郭其祥
合成化学 2024年2期
廖 健, 王煜洋, 郭其祥
(西南大学 化学化工学院,重庆 400715)
近年来,手性醛催化剂在手性胺合成化学中起到了越来越重要的作用。例如,手性醛可催化烯丙基胺与羟胺的不对称氢胺化反应[1],氨甲基类化合物的不对称ɑ-官能团化反应[2-9],以及ɑ-羰基羧酸的转胺化反应[10]。目前所报道的手性醛催化剂可分为3大类(图1):一是BEAUCHEMIN课题组[1]所报道的手性脂肪醛催化剂1;二是GUO课题组所报道的含有联萘骨架的轴手性醛催化剂2[2]、3[3];三是ZHAO课题组所报道的手性吡哆醛类催化剂4[10]、5[4]。其中,GUO课题组所报道的手性醛催化剂3在催化甘氨酸衍生物与不饱和共轭烯酮的加成环化[4],以及氨基酸酯与对亚甲基苯醌的1,6-共轭加成反应[5]中均取得了极其优异的结果。

图1 已报道的手性醛催化剂Figure 1 Reported chiral aldehyde catalysts
目前已报道的合成手性醛催化剂3的路线如图2所示[3]。该路线经9步反应,以约10%的总收率得到手性醛催化剂3,总收率较低。其主要原因为:由11经DIBAL-H还原合成12时收率仅为50%。且在该合成路线中,由于带吸电子取代基的芳基格试剂制备比较困难,因此未能得到具有吸电子芳基取代的手性醛催化剂3,这在一定程度上限制了手性醛催化剂3的生成。因此,对手性醛催化剂3的合成路线进行改进十分必要。

图2 已报道的手性醛催化剂3的合成路线Figure 2 Reported synthesis route of chiral aldehyde catalysts 3
基于此,为了发展一种能够以较高收率获得不同取代基取代的手性醛催化剂3,且适用于大量合成的合成方法。因此,根据相关文献报道[3,11-15],重新设计了手性醛催化剂3的合成路线(图3)。 (S)-BINOL与对甲苯磺酰氯以及三氟甲磺酸酐进行磺酰化反应[11]得到(S)-1,1′-联萘-2′-((三氟甲基磺酰基)氧基)-2-((4-甲基苯基磺酰基)氧基)(13),13在醋酸钯的催化下进行插羰反应[11],得到(S)-1,1′-联萘-2′-((4-甲基苯基磺酰基)氧基)-2-乙氧羰基(14)。14发生镍催化的Suzuki偶联反应[11-12]得到(S)-1,1′-联萘-2′-芳基-2-乙氧羰基(15),15在Mg(TMP)2[3,13]作用下,与硼酸三甲酯反应,经盐酸水解得到(S)-(2-(乙氧羰基)-2′-芳基-[1,1′-联萘]-3-硼酸(16)。16在双氧水作用下得到(S)-1,1′-联萘-2′-芳基-3-羟基-2-甲酸乙酯(17),17经溴甲基甲基醚保护[14]得到(S)-1,1′-联萘-2′-芳基-3-(甲氧基甲氧基)-2-甲酸乙酯(18)。18经氢化铝锂还原得到(S)-1,1′-联萘-2′-芳基-3-(甲氧基甲氧基)-2-甲醇(19),19发生戴斯马丁氧化反应得到(S)-1,1′-联萘-2′-芳基-3-(甲氧基甲氧基)-2-甲醛(20)。20经盐酸作用脱除甲氧基甲基得到12,12经N-溴化琥珀酰亚胺(NBS)溴化[15]得到手性醛催化剂3。
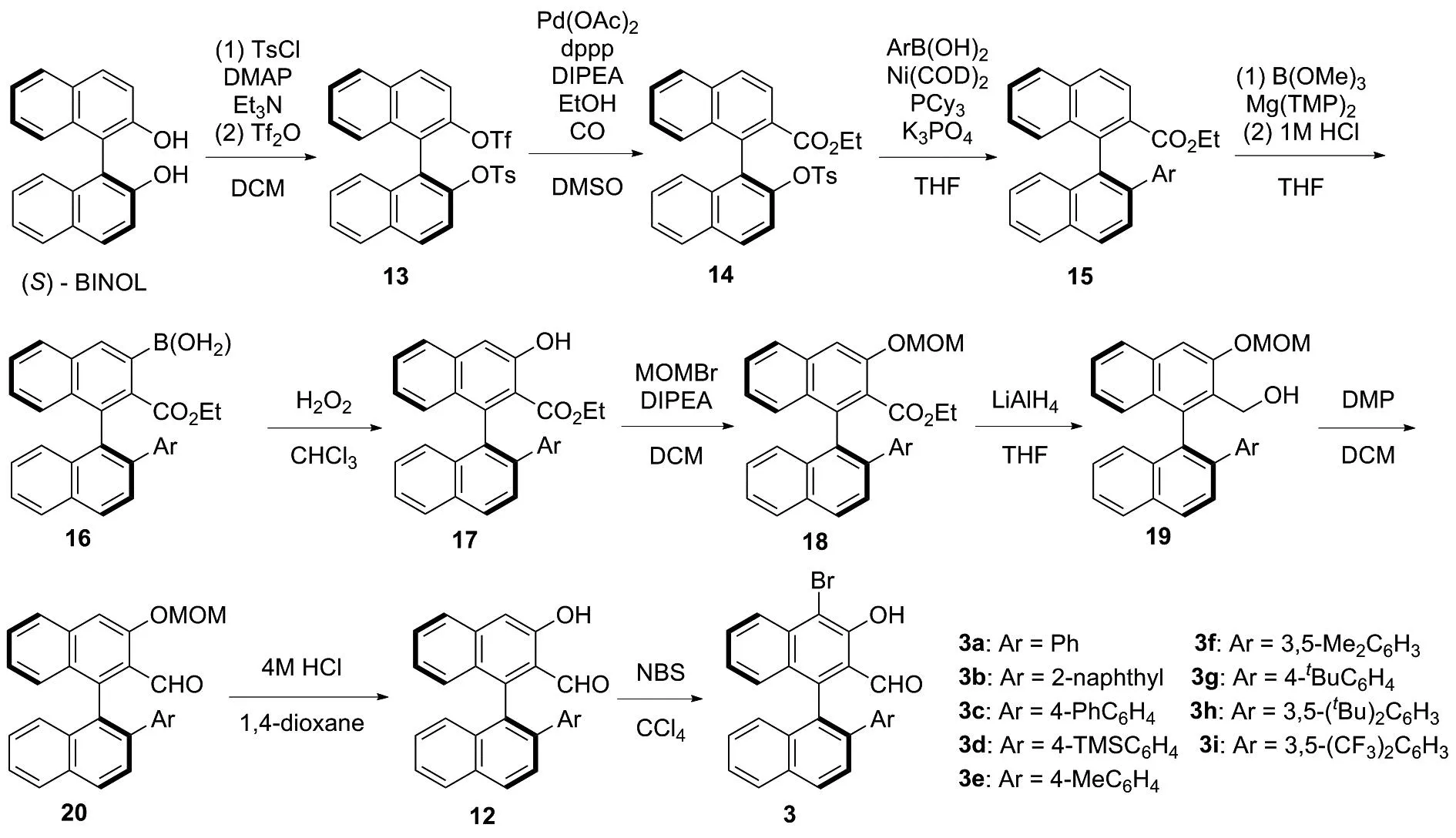
图3 手性醛催化剂3的合成新路线Figure 3 New synthesis route of chiral aldehyde catalysts 3
1 实验部分
1.1 仪器与试剂
卓的-ZD90型电热数字显示熔点仪; Perkin-Elmer-341型自动旋光仪; Bruker-400/600 MHz型核磁共振仪; Bruker-impact II型高分辨率质谱仪。
所用试剂均为分析纯。
1.2 合成
(1)13的合成
根据文献[11]合成13。
(2)14的合成

(3)15a~15i的合成(以15b为例)
在手套箱中,于干燥的48 mL耐压瓶中加入双(1,5-环辛二烯)镍(0.06 g, 0.23 mmol)、三环己基膦(0.25 g, 0.90 mmol)和四氢呋喃(5 mL),并用磁力搅拌子搅拌5 min。加入14(1.48 g, 3.00 mmol)、 2-萘硼酸(1.55 g, 9.00 mmol)、磷酸三钾(1.91 g, 9.00 mmol)和四氢呋喃(10 mL),并于80 ℃下搅拌48 h。向反应体系中加水稀释,混合物用乙酸乙酯(3×15 mL)萃取,合并有机相。有机相经无水硫酸钠干燥后减压浓缩,经柱层析得15b(1.06 g)。经由相同方法合成15a~15i。
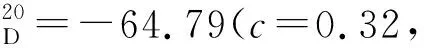





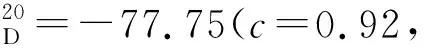


(4)16a~16i的合成(以16b为例)
在干燥的100 mL三颈烧瓶中加入15b(1.06 g, 2.30 mmol),并用磁力搅拌子搅拌,氮气置换3次,加入四氢呋喃(10 mL)。将体系冷却至-78 ℃,滴加Mg(TMP)2(26.00 mL, 11.50 mmol),搅拌5 min后升至室温搅拌5 h。然后再将体系冷却至-78 ℃,滴加硼酸三甲酯(1.50 mL, 13.80 mmol),于该温度下搅拌30 min后升至室温搅拌24 h。将体系冷却至0 ℃,滴加15 mL 1M盐酸,升至室温继续搅拌2 h。混合物用乙酸乙酯(3×15 mL)萃取,合并有机相。有机相经无水硫酸钠干燥后减压浓缩得到16b,无需进一步纯化,直接进行下一步投料。经由相同方法合成16a~16i。
(5)17a~17i的合成(以17b为例)
在50 mL圆底烧瓶中加入16b、氯仿(10 mL),并用磁力搅拌子搅拌。将体系冷却至0 ℃,滴加过氧化氢(质量分数为30%, 2.30 mL, 23.00 mmol),升至室温搅拌10 h,再次将体系冷却至0 ℃,加入饱和亚硫酸钠溶液淬灭反应。混合物用二氯甲烷(3×15 mL)萃取,合并有机相。有机相经无水硫酸钠干燥后减压浓缩得到17b,无需进一步纯化,直接进行下一步投料。经由相同方法合成17a~17i。
(6)18a~18i的合成(以18b为例)
在50 mL圆底烧瓶中加入17b、N,N-二异丙基乙胺(1.60 mL, 9.20 mmol),并用磁力搅拌子搅拌,加入二氯甲烷(10 mL),将体系冷却至0 ℃,滴加溴甲基甲基醚(0.30 mL, 3.45 mmol)。升至室温搅拌1 h,再次将体系冷却至0 ℃,加水淬灭反应。混合物用二氯甲烷(3×15 mL)萃取,合并有机相。有机相经无水硫酸钠干燥后减压浓缩,经柱层析得18b(0.85 g)。经由相同方法合成18a~18i。
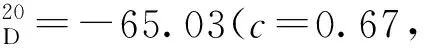


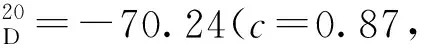



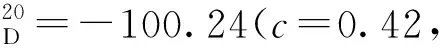
(7)19a~19i的合成(以19b为例)
在100 mL圆底烧瓶中加入18b(1.85 g, 1.67 mmol),并用磁力搅拌子搅拌。加入四氢呋喃(15 mL),将体系冷却至0 ℃,缓慢加入氢化铝锂(0.13 g, 3.34 mmol)。升至室温搅拌1 h,再次将体系冷却至0 ℃,加水淬灭反应。混合物用乙酸乙酯(3×15 mL)萃取,合并有机相。有机相经无水硫酸钠干燥后减压浓缩得到19b,无需进一步纯化,直接进行下一步投料。经由相同方法合成19a~19i。
(8)20a~20i的合成(以20b为例)
在100 mL圆底烧瓶中加入19b,并用磁力搅拌子搅拌,加入二氯甲烷(10 mL),将体系冷却至0 ℃,缓慢加入戴斯-马丁氧化剂(DMP)(2.12 g, 5.01 mmol)。升至室温搅拌2 h,再次将体系冷却至0 ℃,加入饱和亚硫酸钠溶液淬灭反应。混合物用二氯甲烷(3×15 mL)萃取,合并有机相。有机相经无水硫酸钠干燥后减压浓缩得到20b,无需进一步纯化,直接进行下一步投料。经由相同方法合成20a~20i。
(9)12a~12i的合成(以12b为例)
在50 mL圆底烧瓶中加入20b,并用磁力搅拌子搅拌,加入1,4-二氧六环(15 mL),将体系冷却至0 ℃,加入4 M盐酸(0.90 mL, 3.34 mmol)。 60 ℃下搅拌2 h,经减压浓缩后加水稀释,混合物用乙酸乙酯(3×15 mL)萃取,合并有机相。有机相经无水硫酸钠干燥后减压浓缩,经柱层析得12b(0.60 g)。经由相同方法合成12a~12i。
12a:黄色固体,收率83%;1H NMR(400 MHz, CDCl3)δ: 10.84(s, 1H), 9.47(s, 1H), 8.02(d,J=8.0 Hz, 1H), 7.91(d,J=8.0 Hz, 1H), 7.64~7.56(m, 2H), 7.44~7.37(m, 2H), 7.27~7.23(m, 2H), 7.20~7.16(m, 1H), 7.08~6.93(m, 5H), 6.90~6.86(m, 2H);13C NMR(101 MHz, CDCl3)δ: 197.4, 156.4, 146.4, 140.9, 140.8, 138.0, 133.4, 132.4, 129.9, 129.8, 129.4, 128.5, 128.4, 128.1, 128.0, 127.9, 127.8, 127.3, 127.2, 127.0, 126.8, 126.3, 124.5, 119.9, 112.1。
12b:黄色固体,收率85%;1H NMR(400 MHz, CDCl3)δ: 10.76(s, 1H), 9.53(s, 1H), 8.06(d,J=8.0 Hz, 1H), 7.94(d,J=8.0 Hz, 1H), 7.70~7.65(m, 1H), 7.63~7.57(m, 2H), 7.48~7.38(m, 5H), 7.35~7.24(m, 4H), 7.18~7.14(m, 1H), 7.13~7.04(m, 2H), 6.98~6.93(m, 1H);13C NMR(101 MHz, CDCl3)δ: 197.3, 156.4, 146.4, 140.8, 138.4, 138.0, 133.5, 132.9, 132.5, 132.2, 130.2, 129.9, 129.5, 128.5, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.5, 127.3, 127.0, 126.8, 126.4, 126.3, 126.2, 126.1, 124.6, 120.0, 112.2。

12d:黄色固体,收率74%;1H NMR(400 MHz, CDCl3)δ: 10.87(s, 1H), 9.47(s, 1H), 8.01(d,J=8.0 Hz, 1H), 7.90(d,J=8.0 Hz, 1H), 7.64(d,J=8.0 Hz, 1H), 7.57(d,J=8.0 Hz, 1H), 7.45~7.38(m, 2H), 7.28~7.19(m, 3H), 7.14~7.10(m, 2H), 7.09~7.03(m, 1H), 6.99(d,J=8.0 Hz, 1H), 6.88(d,J=8.0 Hz, 2H), 0.08(s, 9H);13C NMR(101 MHz, CDCl3)δ: 197.4, 156.5, 146.5, 141.1, 140.8, 139.3, 138.0, 133.5, 133.1, 132.4, 129.8, 129.7, 129.4, 128.6, 128.1, 128.0, 127.9, 127.8, 127.2, 127.0, 126.8, 126.3, 124.5, 119.9, 112.1, -1.2。

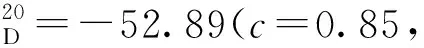
12g:黄色固体,收率79%;1H NMR(400 MHz, CDCl3)δ: 10.86(s, 1H), 9.44(d,J=8.0 Hz, 1H), 7.99(d,J=8.0 Hz, 1H), 7.88(d,J=8.0 Hz, 1H), 7.63(d,J=8.0 Hz, 1H), 7.57(d,J=12.0 Hz, 1H), 7.43~7.36(m, 2H), 7.27~7.15(m, 3H), 7.07~7.01(m, 1H), 7.00~6.95(m, 3H), 6.83~6.78(m, 2H), 1.10(s, 9H);13C NMR(101 MHz, CDCl3)δ: 197.5, 156.5, 150.0, 146.7, 140.8, 137.9, 137.7, 133.5, 132.3, 129.8, 129.7, 129.3, 128.6, 128.3, 128.2, 128.1, 128.0, 127.1, 127.0, 126.7, 126.1, 125.0, 124.5, 119.9, 112.0, 34.4, 31.1。


(10)3a~3i的合成(以3b为例)
在50 mL圆底烧瓶中加入12b(0.60 g, 1.40 mmol),并用磁力搅拌子搅拌。然后再加入四氯化碳(14 mL),将体系冷却至0 ℃,缓慢加入NBS(0.37 g, 2.10 mmol),升至室温搅拌8 h,再次将体系冷却至0 ℃,滴加饱和亚硫酸钠溶液淬灭反应。混合物用二氯甲烷(3×15 mL)萃取,合并有机相。有机相经无水硫酸钠干燥后减压浓缩,经柱层析得3b(0.63 g)。经由相同方法合成3a~3i。
3a:黄色固体,收率96%;1H NMR(400 MHz, CDCl3)δ: 11.72(s, 1H), 9.50(s, 1H), 8.26(d,J=8.0 Hz, 1H), 8.11(d,J=8.0 Hz, 1H), 7.99(d,J=8.0 Hz, 1H), 7.69~7.61(m, 2H), 7.51(t,J=8.0 Hz, 1H), 7.39(d,J=8.0 Hz, 1H), 7.34~7.30(m, 1H), 7.23~7.19(m, 1H), 7.11~7.03(m, 4H), 6.96~6.94(m, 2H);13C NMR(151 MHz, CDCl3)δ: 196.8, 153.3, 146.1, 141.1, 140.6, 136.1, 133.3, 132.4, 131.2, 129.7, 129.2, 129.1, 128.4, 128.3, 128.2, 128.1, 127.9, 127.5, 127.4, 126.6, 126.4, 126.3, 125.1, 119.9, 107.3。
3b:黄色固体,收率90%;1H NMR(400 MHz, CDCl3)δ: 11.65(s, 1H), 9.56(s, 1H), 8.25(d,J=8.0 Hz, 1H), 8.14(d,J=8.0 Hz, 1H), 8.01(d,J=8.0 Hz, 1H), 7.74(d,J=8.0 Hz, 1H), 7.68~7.62(m, 2H), 7.54~7.46(m, 5H), 7.39~7.32(m, 3H), 7.27~7.23(m 1H), 7.08(d,J=8.0 Hz, 1H), 7.02~6.99(m, 1H);13C NMR(151 MHz, CDCl3)δ: 196.8, 153.3, 146.1, 141.0, 138.2, 136.1, 133.4, 132.9, 132.5, 132.2, 131.3, 129.8, 129.4, 129.2, 128.4, 128.3, 128.1, 128.0, 127.9, 127.8, 127.6, 126.6, 126.5, 126.4, 126.3, 126.2, 126.1, 125.2, 119.9, 107.4。

3d:黄色固体,收率91%;1H NMR(600 MHz, CDCl3)δ: 11.59(s, 1H), 9.32(s, 1H), 8.10(d,J=6.0 Hz, 1H), 7.92(d,J=6.0 Hz, 1H), 7.80(d,J=6.0 Hz, 1H), 7.49~7.44(m, 2H), 7.35~7.30(m, 1H), 7.24(d,J=6.0 Hz, 1H), 7.13(t,J=6.0 Hz, 1H), 7.06~7.02(m, 3H), 6.84(d,J=6.0 Hz, 1H), 6.77(d,J=6.0 Hz, 2H), 0.00(s, 9H);13C NMR(151 MHz, CDCl3)δ: 196.9, 153.3, 146.2, 141.0, 140.8, 139.6, 136.1, 133.4, 133.2, 132.4, 131.3, 129.7, 129.3, 129.0, 128.5, 128.2, 128.0, 127.7, 127.4, 126.6, 126.4, 126.3, 125.1, 119.9, 107.3, -1.3。

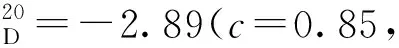
3g:黄色固体,收率92%;1H NMR(400 MHz, CDCl3)δ: 11.77(s, 1H), 9.48(s, 1H), 8.28(d,J=8.0 Hz, 1H), 8.09(d,J=8.0 Hz, 1H), 7.98(d,J=8.0 Hz, 1H), 7.69~7.61(m, 2H), 7.52~7.47(m, 1H), 7.43~7.38(m, 1H), 7.32~7.27(m, 1H), 7.24~7.19(m, 1H), 7.11~7.05(m, 2H), 7.03~6.98(m, 1H), 6.91~6.87(m, 2H), 1.20(s, 9H);13C NMR(101 MHz, CDCl3)δ: 197.0, 153.3, 150.2, 146.4, 140.9, 137.5, 136.0, 133.4, 132.3, 131.3, 129.6, 129.3, 128.9, 128.5, 128.2, 128.1, 127.4, 126.6, 126.4, 126.3, 125.2, 125.1, 119.8, 107.2, 34.4, 31.1。

2 结果与讨论
2.1 放大反应
该合成方法中,由(S)-BINOL合成13,如图4(a)所示,可在50 mmol的规模上得到90%的收率。13可以在20 mmol的规模上以71%的收率合成14,如图4(b)所示。从14(10.00 mmol)出发合成3a,如图4(c)所示,经偶联反应、硼化、氧化、甲氧基甲基保护、还原、氧化、脱保护以及溴代8步反应能以29%的总收率得到目标产物。
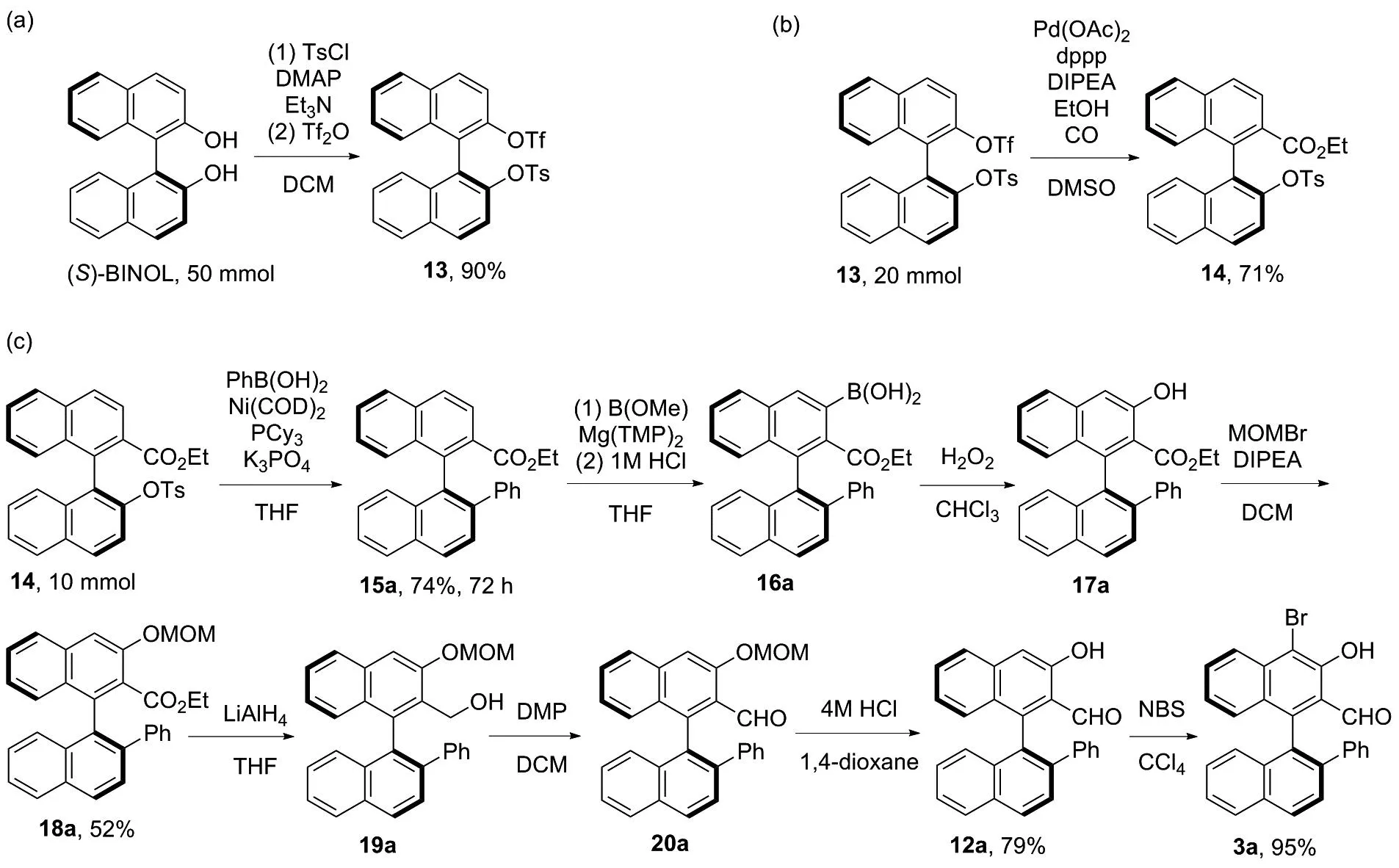
图4 手性醛3a的克级规模合成Figure 4 Gram-scale synthesis for chiral aldehyde 3a
2.2 烷氧基对17合成的影响
在由13合成17的过程中,烷氧基对于17收率有较大的影响,因此在其它条件不变的情况下,考察了烷氧基对17收率的影响(图5,表1)。当烷氧基为甲氧基时,有利于插羰反应以及偶联反应,能以29%的总收率得到目标产物(Entry 1)。而使用正丙基氧基时,则是有利于在酯基邻位引入羟基,能以22%的总收率得到目标产物(Entry 3)。当选用乙氧基时,各步反应都能有中等的收率,能以36%的总收率得到目标产物(Entry 2)。因此,乙氧基为最优的烷氧基。

表1 烷氧基对17合成中收率的影响Table 1 Effect of alkoxy group on the synthesis of the yield of 17
2.3 17合成12的条件筛选
在由17合成12的过程中,对条件进行了一系列筛选。首先选用DIBAL-H对17b直接进行还原,能以36%收率得到12b,如图6(a)所示。之后尝试选用氢化铝锂对17b进行还原,以63%收率得到(S)-1,1′-联萘-2′-(2-萘基)-3-羟基-2-甲醇(21),再对21进行Parikh-Doering氧化[16],能以43%收率进行氧化,以总收率27%得到12b,如图6(b)所示。此外,尝试使用溴甲基甲基醚对17b酚羟基进行保护,能以90%收率得到18b,再使用氢化铝锂对18b进行还原,能以83%收率得到19b。之后使用戴斯-马丁氧化剂对19b进行氧化,能以91%收率得到20b,使用盐酸能以90%收率对保护基进行脱除,以4步总收率61%得到12b,如图6(c)所示。因此,选用收率最高的路径,经保护、还原、氧化和脱保护4步合成12b。
2.4 氧化剂的筛选
在合成20的过程中,发现氧化剂的种类会对反应收率产生影响。因此在其它条件不变的情况下,考察了氧化剂类型对反应的影响。在使用DMP时,能以91%收率得到目标产物(表2, Entry 1),相较于氯铬酸吡啶盐(PCC)效果更好(80%, Entry 2),因此选用DMP作氧化剂。

表2 氧化剂的筛选Table 2 Selection of oxidant
本文报道了手性醛催化剂(3)合成路线的优化方法。相较于文献中已报道的路线,该路线能以大于20%的总收率合成大部分2′-位具有更多类型取代基的3;能以12%的总收率合成在2′-位带有强吸电子基团的芳基(3,5-二(三氟甲基)苯基)的3,这对于多样性3的获取具有一定的积极作用。该合成路线避免了使用具有强腐蚀性的溴素作为溴源,而是选用对环境更加友好的NBS作为溴源进行溴化反应,且该方法能放大到10 mmol规模来进行,具有一定应用前景。
猜你喜欢
分子催化(2022年1期)2022-11-02
硫酸工业(2020年2期)2020-04-16
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
合成化学(2015年2期)2016-01-17
动物营养学报(2015年9期)2016-01-07
化工进展(2015年6期)2015-11-13
中国塑料(2015年10期)2015-10-14
中国洗涤用品工业(2015年4期)2015-02-28
郑州大学学报(理学版)(2014年3期)2014-03-01
