
基于临床试验评价新型双重食欲素受体拮抗剂类抗失眠药物daridorexant 的有效性及安全性
2024-04-09 01:56陶云松贾元威
中国合理用药探索 2024年2期
陶云松,贾元威
1 芜湖市第二人民医院,芜湖 241001;2 皖南医学院弋矶山医院,芜湖 241001
双重食欲素受体拮抗剂(dual orexin receptor antagonists,DORAs)是一类通过同时拮抗食欲素1 型受体(orexin-1 receptor,OX1R)和OX2R 来改善失眠症状的新型药物。食欲素(orexin)系统又称下丘脑分泌素系统,是控制和协调所有促觉醒区域活动的神经系统,可维持觉醒的稳定性,并根据各种生理过程(如摄食、奖赏和应激过程)调节警觉性水平[1-2]。食欲素系统由位于下丘脑外侧的食欲素神经元组成,这些神经元能分泌食欲素A 和食欲素B 2 种神经肽。食欲素在脑细胞外和脑脊液中的浓度呈现昼夜节律变化:在觉醒期间升高,在睡眠期间迅速下降。食欲素A 和食欲素B 通过与OX1R 和OX2R 结合,对觉醒-睡眠产生不同影响,OX1R 主要参与抑制快速眼动(rapid eye movement,REM)睡眠,OX2R 主要参与抑制非快速眼动(non-REM)[3-4]睡眠。食欲素受体拮抗剂能阻断食欲素与食欲素受体结合,可以抑制过度的促觉醒信号,抵消失眠患者的脑过度活跃状态,从而帮助患者改善失眠症状[5]。
首个进入临床研究的DORAs 是almorexant,但未成功上市[6]。首个成功上市的DORAs 是suvorexant[7]。Lemborexant 是第2 个上市的DORAs,于2019 年在美国上市,2020 年在日本上市,用于治疗失眠[8-9]。Daridorexant 于2022年1 月在美国上市,用于治疗成人失眠患者的入睡和睡眠维持困难[10]。DORAs 药物的全球研发与上市情况见表1。目前,daridorexant 已在欧盟、日本、英国和加拿大等国家或地区获得上市许可,也是目前为止首个获批用于治疗慢性失眠且可改善日间功能的药物,相关药物专利均已超过20 年保护期,同品种未在我国上市。
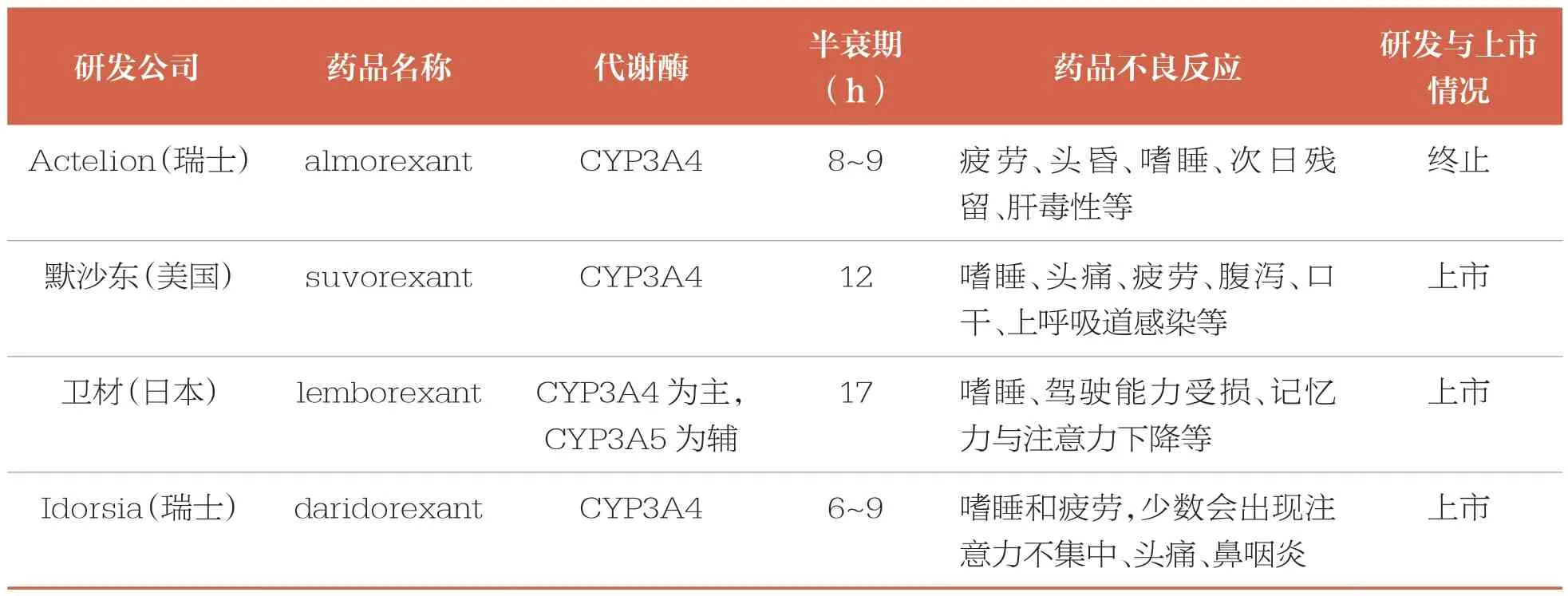
表1 DORAs 药物的全球研发与上市情况
新药临床有效性和安全性评价是新药剂量-暴露-效应关系及获益-风险评估的重要内容。药物临床试验中,评价药物的有效性主要通过有效性指标的观测和评价来实现。申报上市申请时的安全性评价是基于上市前所有临床试验,总结其在新药安全性方面的重要发现及相关证据,以及评估新药的总体安全性特征。本文基于daridorexant 已经开展的Ⅰ~Ⅲ期临床试验结果,对其临床应用的安全性及有效性进行分析,以期对daridorexant 后续研发及临床应用提供参考。
1 Ⅰ期临床研究
1.1 单次给药剂量递增(single ascendingdose,SAD)药动学研究
SAD 药动学研究探索了具有药理学活性暴露量水平的剂量范围,同时考察了暴露量与安全性和耐受性的相关性。在首次人体研究(NCT02919319)中[11],40 例男性健康志愿者分为5 组, 每组8 例,其中6 例在晨起后接受ACT-541468(daridorexant 活性成分)5~200mg 的单次给药,2 例接受安慰剂。药动学研究显示,ACT-541468的绝对生物利用度为62%,分布容积为31L;与血浆蛋白的结合率达99.7%,血液/血浆比为0.64。口服给药后,ACT-541468 单次给药5~50mg 在1~2h 内达到血浆峰浓度;单次给药100mg 达峰时间为2.5h;单次给药200mg 达峰时间为2.8h。其血浆暴露量在剂量25~50mg 范围内与剂量呈线性关系。当健康受试者进食高脂高热量餐后,ACT-541468 50mg 的达峰时间延迟1.3h,峰浓度降低16%,但总暴露量不受影响。ACT-541468 5~50mg 在成人中的半衰期约为6h[11]。后续研究发现,其在老年人中的半衰期约为10h[10]。
ACT-541468 在25mg 及更高剂量给药后1h内可引起中枢神经系统相关效应,包括警觉性、注意力、视觉运动协调和姿势稳定性的降低。这些效应在给药后3~10h 内回到基线水平,其中,25mg剂量给药后在3~6h 内回到基线水平;50mg 剂量给药后在6~8h 内回到基线;100mg 剂量的最大效应出现在给药后1.5h,大多数受试者在8h 内回到基线;而最大单次给药剂量为200mg 时,最大效应出现在给药后2h,大多数受试者在10h 内回到基线。单剂量给药研究中出现的不良事件均在轻度到中度,最常见不良事件为嗜睡(在安慰剂组和试验组均有出现)、疲劳、注意力障碍、头痛,没有出现因不良事件而退出试验的情况。最高试验剂量下(200mg,早晨给药)的6 例健康受试者中有4 例报告了轻度肌无力,发生在给药后45min~2.5h,持续时间最短者为1min,最长者达3h,研究者将其评估为与研究给药相关。研究者认为这些病例中仅1 例可能为猝倒症样事件[11]。
1.2 多次给药剂量递增(multiple ascendingdose,MAD)药动学研究
MAD 药动学研究基于目标适应症特点、研究药物的非临床研究数据以及前期SAD 研究数据等,关注预期浓度范围内剂量-暴露比例关系特征、消除半衰期、药效持续时间、蓄积情况等。在daridorexant 的多次给药研究(NCT02571855)中,首次服用ACT-541468 后1h 出现相关效应,最大效应约在服药后2h 出现,8~10h 内回到基线水平[12]。在多次给药后的第5 天(d5),在服药后1h 观察到最大效应,在给药后10h 内回至基线水平[12]。单次和多次给药均显示,药效学效应的强度和持续时间随剂量增加而增加。
ACT-541468 多次给药后的药动学特征与单次给药后相似,未观察到明显的药物蓄积[12],且具有良好的药动学和药效学特性,吸收和消除快速,多项研究认为这可能是其次日残留效应罕见的主要原因[13-16]。
在MAD 研究中,有3 例受试者报告了轻度肌无力:10mg 剂量组1 例和75mg 剂量组2 例受试者。经研究者判断,这些事件均不是猝倒症样事件[12]。在I 期研究中,daridorexant 75mg 剂量组在多次给药后有发生睡眠性瘫痪的报告。睡眠性瘫痪是指在睡眠-觉醒转换过程中出现无法移动或说话的情况,持续通常几秒钟,也可能长达几分钟,有时伴有催眠幻觉,该不良反应与已知的食欲素受体拮抗剂药理作用一致[12]。
1.3 药物代谢、药物相互作用及食物的影响
Daridorexant 经CYP3A4 代谢的比例达89%,其他CYP 酶的代谢作用均不明显。在人体内,daridorexant 的主要代谢物为M1、M3 和M10,这些代谢物对daridorexant 的药理作用没有影响。Daridorexant 的排泄主要通过粪便(约57%),其次是尿液(约28%)。在尿液和粪便中,原型药物的含量很低[17]。
中度和强效CYP3A4 抑制剂会抑制daridorexant的代谢,导致其血药浓度升高,增加药物中毒的可能性。如与地尔硫 (中度CYP3A4 抑制剂)合用时,daridorexant 的药时曲线下面积(area under the plasma concentration-time curve,AUC)可增加至240%;与强效CYP3A4 抑制剂合用时,daridorexant的AUC可增加至400%[18]。相反,中度和强效CYP3A4 诱导剂会促进daridorexant 的代谢,导致其血药浓度降低,影响其疗效。如与依法韦伦(中度CYP3A4 诱导剂)合用时,daridorexant 的AUC降低35%左右;与利福平(强效CYP3A4 诱导剂)合用时,daridorexant 的AUC下降50%以上[19]。相关研究提示daridorexant 应避免与中度/强效诱导剂及强效CYP3A4 抑制剂联用,如确需与中度CYP3A4 抑制剂联用时,最大剂量不应超过25mg。另外,daridorexant 与其他中枢神经系统抑制剂联用时,也应谨慎调整药物剂量,以防止出现药品不良反应。精神科常见的CYP3A4 抑制剂有安非他酮、氟伏沙明等,CYP3A4诱导剂有奥卡西平、卡马西平等。说明书推荐剂量为25~50mg,随餐或餐后服用daridorexant 会延迟入睡时间,因此建议每晚睡前半小时空腹服用。此外,与乙醇合用会延长daridorexant 的达峰时间,并增加精神运动表现的损害[20],服用daridorexant 期间应避免饮酒。
1.4 肝肾功能不全人群的临床研究
轻度肝功能损害(Child-Pugh A)患者服用daridorexant 25mg 剂量后,游离daridorexant 的暴露量与健康人群相当。中度肝功能损害(Child-Pugh B)患者服用daridorexant 25mg 剂量后,游离daridorexant 的AUC和半衰期分别是健康人群的1.6 倍和2.1 倍。因此,参照药品说明书,对于中度肝功能损伤的患者,daridorexant 的最大剂量应调整为25mg/晚;对于重度肝功能损伤的患者,不建议使用该药[17,21]。
重度肾功能损害患者服用daridorexant 25mg剂量后,其药动学参数与健康人相似。提示肾功能不全对daridorexant 的药效影响不大[22]。
2 Ⅱ期临床研究
针对daridorexant 的有效性,已完成的多项Ⅱ期临床研究[13]均在符合《精神障碍诊断与统计手册(第5 版)》(DiagnosticandStatistical Manual5thEdition,DSM-5)标准的失眠患者中进行。其中研究NCT 02839200(ClinicalTrials.gov)/EudraCT(2016-000826-21)(以下简称研究201)是平行组设计,在6 个国家的38 个研究中心医院和睡眠中心开展,该研究筛选的1005例受试者中,纳入了360 例成人患者(18~64 岁),治疗期为29 天[13]。
研究201 将患有失眠症的成人(≤64 岁)随机分组,每日接受口服安慰剂、daridorexant(5、10、25mg 或50mg)剂量或唑吡坦10mg 剂量治疗30 天。主要疗效指标是入睡后觉醒时间(waketime after sleep onset,WASO),即维持睡眠能力的指标。主要研究终点是从基线到第1 天和第2天在中心多导睡眠图(polysomnography,PSG)监护下的睡眠及觉醒参数的变化;次要研究终点是从基线到第1 天和第2 天睡眠潜伏期(latency to persistent sleep,LPS)的变化,以及从基线到第4 周睡眠开始后主观睡眠潜伏期和主观睡眠参数的变化。其中LPS 是反映入睡是否困难的指标。依据受试者每天早晨和晚上记录的日记评估主观睡眠参数,即WASO、LPS 和主观总睡眠持续时间(subjective total sleep time,sTST)。 在基线到4 周治疗期间,依据日记记录每7 天取平均值。此外,每日早晨和晚上以视觉模拟量表评分法(visual analogue score,VAS)评价受试者主观感觉的晨起嗜睡、日间警觉性、日间功能、次日残留效应等。
在筛选阶段,受试者至少连续7 天完成睡眠日记评估(sleep diary questionnaire,SDQ),以评估基线睡眠特征。在连续2 个单盲安慰剂治疗的夜间记录PSG。受试者需要满足以下PSG 标准才能随机分组:①2 个夜晚的平均WASO≥30min。②LPS≥30min。③TST<420min。受试者失眠严重程度指数(insomnia severity index,ISI)至少达到15 分。排除标准包括有自杀行为、自杀意念、不稳定医学状况或严重医学疾病的受试者,其他排除标准如需要药物治疗的失眠以外的持续睡眠障碍、每小时10 次以上的呼吸暂停/通气不足等。
在研究201 中,将359 例受试者(64%为女性)随机分组。从基线到使用daridorexant 的第2天,LPS 和WASO 指标改善存在显著的剂量-效应关系(P<0.001),且一直持续到第28 天和第29 天(P=0.050 和P=0.042)。主观睡眠参数及睡眠质量同样体现剂量-效应关系。与此相比,唑吡坦10mg(阳性对照组)改善了LPS 参数,但对WASO 无明显影响,这与先前的报道相符[23]。
另外一项Ⅱ期研究NCT02841709(ClinicalTrials.gov)/EudraCT (2016-000827-16)(以下简称研究202)在德国和美国的10 个研究中心医院进行[14]。58 例老年(≥65 岁)失眠症患者接受14 天的安慰剂导入后,按拉丁方设计的双盲随机程序依次分为daridorexant 5mg、10mg、25mg 和50mg 剂量组和安慰剂组,每个疗程接受连续2 天PSG 监护的双盲组,然后洗脱5~12 天。在治疗结束后(5 天和30 天),通过电话对参与者进行安全随访。同时也采用量表评估受试者的日间功能、情绪等安全性和耐受性。结果与研究201 类似,daridorexant 剂量依赖性改善了老年受试者睡眠障碍的主要疗效指标和次要疗效指标[14]。
安全性方面,研究201 的结果显示,在接受daridorexant 5mg、10mg、25mg 和50mg 剂量治疗的受试者中,给药后不良事件(treatment emerged adverse event,TEAE)的发生率分别为35%、38%、38%和34%,安慰剂为30%,唑吡坦10 mg 为40%。Daridorexant 组最常见的TEAE 是头痛(10%)、嗜睡(7%)、腹泻和疲劳(5%);治疗组和阳性对照唑吡坦组TEAE 的发生率高于安慰剂组,但无剂量依赖性(药物治疗组中50mg 剂量组的不良事件发生率最低)。在研究202 中,daridorexant 治疗期间TEAE 的发生率较研究201 更低。而各Ⅱ期临床研究中所有TEAE均没有剂量依赖性[13-14]。
3 Ⅲ期临床研究
3.1 有效性评价
2018 年10 月~2021 年2 月,2 项全球确证性Ⅲ期临床研究(NCT03545191,ID-078A301,以下简称研究301)和NCT03575104(ID-078A302,以下简称研究302)[15]在14 个国家的94 家研究中心同时开展,旨在评估daridorexant 治疗失眠障碍的有效性和安全性。相关研究均为多区域、随机、双盲、安慰剂对照研究,纳入了成人(18 岁≤x<65 岁)和老年(≥65 岁)失眠患者,共1854例受试者随机接受daridorexant 或安慰剂,每日1次,晚间给药,持续给药3 个月。2 项Ⅲ期确证性研究的设计相同,主要有效性终点均为第1 个月和第3 个月时WASO 和LPS 较基线的变化。次要终点包括经验证调查问卷测量的患者报告结局,即自我报告的sTST、每天早晨在家记录SDQ 以及日间功能[每天晚上在家评估日间失眠症状和功能影响问卷(nsomnia daytime symptoms and impacts questionnaire,IDSIQ)]。
全球确证性Ⅲ期研究完成第一阶段后,共计1684 例患者完成了12 周双盲治疗和安慰剂对照试验(研究301∶n=847;研究302∶n=837)。在2 项研究中,daridorexant 剂量依赖性改善了WASO 和LPS,并增加了sTST。其中,研究301:daridorexant 25mg 和50mg 剂量组在第1 个月和第3 个月时,相比基线和安慰剂组,均显著降低了WASO 和LPS(P<0.0001,P<0.0005)。 服用daridorexant 50mg 剂量组的患者,1 个月后WASO 平均下降29.0min,安慰剂组下降6.2min;3 个月后WASO 平均下降29.0min,安慰剂组下降11.1 min;服用daridorexant 的患者还显著增加了sTST(P<0.05);IDSIQ 睡眠评分中嗜睡项具有统计学差异。研究302∶daridorexant 25mg 剂量组在第1 个月和第3 个月时,相比基线和安慰剂组,均显著降低了患者的WASO(P<0.0001,P=0.003)和LPS(P<0.03,P<0.005)。服用daridorexant 25mg剂量组的患者,1 个月后WASO 平均下降24.2 min,安慰剂组下降12.6min;3 个月后WASO 平均下降24.3 min,安慰剂组下降14.0 min。服用daridorexant 患者的sTST 显著增加(P<0.0001),但IDSIQ 睡眠评分未见统计学差异[15]。
在全球确证性Ⅲ期研究的第二阶段,完成研究301 和302 的12 周治疗的成人失眠患者,受邀继续参加40 周的双盲扩展研究(NCT03679884,ID-078A303, 以下简称研究303), 以评估daridorexant 的长期安全性和耐受性(主要目的)及其对sTST 和IDSIQ 的影响(次要目的)。在研究303 中,接受daridorexant 治疗的受试者继续使用相同的剂量(10、25mg 或50mg),而接受安慰剂治疗的受试者则以1∶1 的比例重新随机分配为安慰剂组或daridorexant 25mg 剂量组。在研究303 中,共有804 例成人和老年受试者接受了随机分配[142 例(10mg)、270 例(25mg)、137 例(50mg)、128 例(安慰剂)和127 例(前安慰剂/daridorexant 25mg)][16]。
研究303 是一项全球性的长期研究,旨在评估失眠药物daridorexant 的安全性、夜间疗效,以及对日间功能的影响。该研究包括了连续52 周的夜间治疗(其中前12 周为关键期),并纳入了成人和老年失眠患者。在整个治疗期间,daridorexant 表现出良好的安全性和耐受性,没有出现新的药物安全警戒信号,也没有发现身体依赖、耐受性降低或反跳现象,这与前12 周的研究结果一致。在探索性的疗效分析中,基于患者自评的主观终点sTST和IDSIQ 评分,daridorexant 能够持续改善患者的总睡眠时间和日间功能,其中50mg 剂量的效果最为显著[16]。
在日本完成的Ⅲ期临床研究ID-078A304(JRCT2031200452,以下简称研究304)是一项在成人(18 岁≤x<65 岁)和老年(年龄≥65岁)受试者中展开的多中心、双盲、随机、安慰剂对照、平行组、确证性研究,治疗持续时间为28 天。该项研究中,共有490 例受试者被随机分配至daridorexant 25mg 剂量组(n=163)、daridorexant 50mg 剂量组(n=163)或安慰剂组(n=164)。该研究的主要有效性目标是评估daridorexant 50mg 剂量对主观睡眠参数的影响。在第4 周时,daridorexant 50mg 剂量组和25mg剂量组在sTST 和LPS 方面均表现出显著改善,其中50mg 剂量组的效应优于25mg 剂量组。
在日本完成的第2 项Ⅲ期临床研究ID-078A305(JapicCTI205444,以下简称研究305)是一项针对成人(18 岁≤x<65 岁)和老年(≥65岁)失眠患者的多中心、随机、开放性、长期研究,旨在评估daridorexant 的安全性和有效性。该研究共招募了154 例受试者,随机分配至daridorexant 25mg 剂量组(n=52)或50mg(n=102)剂量组,治疗期为52 周(1 年)。该研究的主要结果显示,daridorexant 在治疗期间有效改善了失眠的主观参数,且安全性良好。
上述验证性Ⅲ期研究中,daridorexant 改善日间功能的作用值得注意[15-16]。与安慰剂相比,daridorexant 在增加sTST 和降低所有IDSIQ 评分方面得到了持续改善。在上述研究中,第1 个月和第3 个月时,在所有治疗组中均观察到IDSIQ评分相对于基线的平均改善。在研究301 中,第1 个月和第3 个月时,50mg 剂量组的降幅均具有统计学差异。在12 周的研究中接受安慰剂治疗的患者,随后的扩展研究中随机分配接受持续40 周daridorexant 25mg 剂量治疗后,sTST 和IDSIQ评分的平均变化也明显改善。在治疗的前3 个月即观察到夜间疗效参数改善,然后是日间功能改善。基于此,推测需要先改善夜间睡眠时间和质量,然后才能改善日间功能。
2022 年4 月,daridorexant 在欧盟上市,用于治疗症状持续存在至少3 个月的成人失眠患者,并且可对日间功能产生作用。随后,daridorexant于2022 年8 月和12 月,分别在英国和瑞士上市,用于改善日间功能的成人失眠症。
3.2 安全性及不良事件
在前述Ⅲ期临床研究[14-16],同时评价了daridorexant 的安全性。在研究301 中,最常见的不良事件为鼻咽炎(50mg 剂量组6%,25mg 剂量组7%,安慰剂组6%)、头痛(50mg 剂量组6%,25mg 剂量组5%,安慰剂组4%)和疲劳(50mg剂量组2%,25mg 剂量组2%,安慰剂组1%),导致停药的不良事件在各组中发生率较低(50mg 剂量组1%,25mg 剂量组2%,安慰剂组3%),严重不良事件在各组中发生率也较低(50mg 剂量组1%,25mg 剂量组1%,安慰剂组2%),且无明显的剂量依赖性。在研究302 中,25mg 组的不良事件发生率与研究301 相似。研究301 和302 汇总后发现,最常报告的不良事件(受试者报告>2%,并且与安慰剂的差异>1%)为头痛,其他不良事件为疲劳、头晕、恶心和嗜睡,在治疗组中发生频率均低于4%[16]。Daridorexant 治疗组和安慰剂组在研究期间发生严重不良事件或导致停药的不良事件发生率总体相当。
完成研究301 和302 的受试者中有804 例继续参加研究303,其中550 例患者(68.4%)完成了40 周的双盲治疗。安慰剂组退出试验的受试者达到23%,daridorexant 治疗组各退出8%~11%,最常见的退出原因是疗效不佳。在40 周双盲期间发生的大多数不良事件的严重程度为轻度/中度(91.2%)。在所有组中,双盲治疗期间最常见的不良事件仍是鼻咽炎。所有其他不良事件包括跌倒、头痛和嗜睡,发生率均<3%,头晕和疲劳在各组中均<2%;未观察到不良事件(adverse event,AE)发生频率或严重程度存在剂量关系的证据;老年受试者中的安全性特征与年轻受试者一致[16]。
在全球确证性研究中,与安慰剂组相比,daridorexant 组中与意外事故和各种损伤(包括跌倒)相关的TEAE 发生率始终较低或相近。总体而言,根据现有临床试验数据,健康受试者及慢性失眠患者口服daridorexant 的安全性及耐受性良好。
在前述Ⅲ期临床研究(确证性和扩展研究)中[14-16],研究者及申办方前瞻性制定了中心临床监测计划,确定密切监测发作性睡病样症状等潜在特别关注的不良事件(adverse event of special interest,AESI),并提交独立安全委员会裁定。AESI 包括下列4 个类别:①与日间过度困倦(excessive daytime sleepiness,EDS)相关的发作性睡病样症状。②与复杂睡眠行为相关的发作性睡病样症状,包括幻觉和睡眠性瘫痪。③与猝倒症事件相关的发作性睡病样症状。④自杀/自残。
研究301 中,接受daridorexant 25mg 剂量和50mg 剂量的受试者中,分别有0.5%和0.3%报告了睡眠性瘫痪(安慰剂组发生率为0),但未观察到严重不良反应。Daridorexant 25mg 剂量组中有6%的受试者报告了入睡前和觉醒前幻觉,但daridorexant 50mg 剂量组及安慰剂组均未发现此类不良事件。没有报道猝倒症或其他复杂睡眠行为的病例,如梦游、梦游驾驶和在不完全清醒的情况下从事其他活动。Daridorexant 治疗后有个别病例报告幻觉,但各种AESI 均未体现剂量依赖性[15]。
在研究303 中,报告了3 例AESI:1 例受试者(25mg)被归类为与EDS 相关的发作性睡病样症状;1 例受试者(50mg)被归类为与复杂睡眠行为相关的发作性睡病样症状,包括幻觉和睡眠性瘫痪,报告为异常做梦;1 例受试者(安慰剂)被归类为自杀/自残。总体而言,全球确证性研究中观察到的发作性睡病样症状的AESI 并不常见,且均未反映典型的发作性睡病表型[16]。
4 其他临床研究
4.1 重要脏器合并症的安全性
既往研究显示[24],DORAs 对患有阻塞性睡眠呼吸暂停(obstructive sleep apnea,OSA)、慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)[25]的高危患者不产生呼吸抑制作用,而苯二氮 类药物有产生呼吸抑制的风险,与阿片类药物联合使用时此风险显著增高[26]。在一项轻度或中度OSA(呼吸暂停-低通气指数为5 至<30 次/h 睡眠)患者的研究中[27],daridorexant未增加呼吸暂停/低通气事件的发生频率,也未导致氧饱和度下降,未影响夜间呼吸功能,与既往研究一致。在睡眠特征方面,daridorexant 对其他睡眠相关终点有良好的调节作用(睡眠效率指数增加和WASO 降低)。在中度COPD(FEV1/FVC 比率≤70%且40%≤FEV1<80%预测值)患者的研究中[28],daridorexant 未导致氧饱和度下降。
心脏毒性方面,基于非临床研究结果,daridorexant Ⅰ~Ⅲ期研究的心电图评估均未显示对QT 间期延长有任何影响。按照ICH E14 进行了全面QTc 研究,给予daridorexant 50mg 和200mg,检测莫西沙星对QT 间期的预期影响。使用浓度-QTc 分析,在daridorexant 血药浓度检测范围内,可以排除超过10ms 的QTc 间期效应。
4.2 生理/心理依赖及药物滥用潜力
与苯二氮 类药物及非苯二氮 类药物相比,已上市的DORAs 基本没有观察到戒断效应[7-9]。在研究201 和202 中,经过30 天的治疗期后,根据苯二氮 类药物戒断症状问卷的结果均未检测到戒断效应[13-14]。
在前述daridorexant Ⅲ期研究中[14-16],无论与其他AE 有无相关,均要求研究者将所有研究治疗的用药过量、误用、滥用或研究治疗错误事件报告为AE。意外服药过量(患者无意中服用了或不确定自己是否多吃了一片药片)是最常报告的与药物滥用可能性有关的AE。以研究303 为例,意外服药过量发生情况为:10mg 剂量组4 例(2.8%);25mg剂量组3 例(1.1%);50mg 剂量组4 例(2.9%);安慰剂组4 例(3.2%)[16]。没有证据表明有戒断症状或反跳性失眠,表明有daridorexant 的滥用可能性很低。在临床试验中未发现与daridorexant 相关的抑郁或抑郁恶化、戒断症状、生理依赖性、反跳性的表现或药物滥用潜力等不良反应。这与临床前关于daridorexant 与滥用相关的中枢神经系统(central nervous system,CNS)靶点亲和力的研究一致[29]。在120 多种酶和放射性配体结合试验中,daridorexant 及其主要代谢物M1、M3 和M10 与已确定的离子通道、受体、转运蛋白和酶的选择无相关脱靶活性。这些包括已知与物质滥用相关的CNS 靶点,如大麻素受体、阿片受体、5-羟色胺受体、γ-氨基丁酸(gamma-aminobutyric acid,GABA)受体、单胺转运蛋白、谷氨酸受体(N-methyl-D-aspartic acid receptor,NMDA)和各种离子通道。
由于daridorexant 具有CNS 活性,美国食品药品监督管理局(Food and Drug Administration,FDA)要求对其进行人类滥用潜力(human abuse potential,HAP)研究,旨在通过评估主观药物喜好和其他与滥用潜力相关的终点,来测试daridorexant 是否会引起滥用或成瘾[30]。一项随机、双盲、双模拟、安慰剂和活性对照的6 阶段交叉研究(NCT03657355)被用来评估daridorexant在娱乐性镇静药物使用者中的滥用潜力,该研究招募了63 例健康的娱乐性镇静药物使用者。主要终点是daridorexant 各剂量组的药物喜好VAS 评分,该评分反映了受试者对药物的主观喜好程度。结果显示,daridorexant 在所有剂量下的药物喜好VAS 评分均大于安慰剂,但只有100mg 和150mg剂量组的daridorexant 与suvorexant(150mg)和唑吡坦(30mg)2 种活性对照药物的药物喜好VAS 评分相当;而50mg 剂量组的daridorexant的药物喜好VAS 评分则显著低于2 种活性对照药物。大多数次要终点也呈现出类似的剂量相关模式。此外,daridorexant 所有剂量的欣快相关AE(即欣快情绪、感觉异常)的发生率(3.0%~5.8%)均低于suvorexant(9.0%)和唑吡坦(20.3%),表明daridorexant 的安全性较好[31]。
4.3 对驾驶车辆和操作机器能力的影响
一项研究使用高灵敏度驾驶模拟器评价了daridorexant 夜间给药对次晨驾驶表现的影响,受试者为60 例50~79 岁且无睡眠障碍的健康人,是一项随机、安慰剂和阳性对照(佐匹克隆)的四交叉研究。检测时间为daridorexant 50mg 和100 mg 给药后9h,分别在首次给药(初始给药)和连续给药4 晚后进行。佐匹克隆7.5mg 作为阳性对照药物。驾驶表现的主要指标是在模拟器中以95km/h的平均速度进行1 h 驾驶测试期间的标准偏差车道位置(standard deviation of lateral position,SDLP)。
SDLP 的测量结果显示,与安慰剂相比,阳性对照药物佐匹克隆在2 个检测时间点均显著影响了模拟驾驶的表现,而daridorexant 在首次给药后的检测时间点影响了模拟驾驶的表现,100mg 剂量的影响更大。但在连续给药4 晚后的检测时间点,daridorexant 的2 种剂量均未对模拟驾驶的表现产生影响。受试者通过VAS自我评定驾驶质量和精力,daridorexant 给药期间的量表结果与SDLP 结果一致,表明受试者能够判断自己的驾驶表现。
5 讨论
目前在临床上广泛应用的抗失眠药物主要有三类:主要作用于GABA 受体的镇静催眠药,包括传统的苯二氮 类药物和非苯二氮 药物[32-33];其次是组胺受体拮抗剂类药物;还有褪黑素受体激动剂类药物[34-35]。然而,上述药物在临床实践中显示出一系列难以克服的不良反应,如滥用风险相对较高、精神障碍、夜间异常睡眠、认知功能障碍等,限制了其在临床上的长期安全应用。以应用最广泛的GABA-A 调节剂为例,通常由于其广泛和过度的神经元通路抑制,产生一些常见的不良反应,例如次日早晨残余嗜睡、跌倒、运动不协调、耐受性和身体依赖,而且不能改善患者认知、困倦、疲乏等日间功能,从而显著限制了GABA-A 调节剂的使用[36-37]。相关指南对此发出警告或不推荐长期使用[38-39]。因此,慢性失眠患者对长期治疗的需求显著而突出[40-41]。
理想的治疗失眠药物应该在短期和长期都能安全有效地改善睡眠的定量和定性指标,包括总睡眠时间、入睡后清醒时间、持续睡眠潜伏期、睡眠质量等,同时也应该可以改善日间功能和总体健康[42]。DORAs 是研究人员基于探索失眠症的发生机制而开发的新一代抗失眠药物,旨在克服现有药物的局限性,具有增加睡眠各个阶段持续时间的特性,同时不改变睡眠结构周期,为患者提供了更为“自然”的睡眠体验。与此同时,其并不引起大脑活动的普遍抑制,避免了对认知功能的干扰或易被唤醒的风险。这种作用机制的另一个优势在于满足了老年人失眠治疗的一些重要需求,特别是对先前存在认知障碍或运动功能障碍的老年人,其在夜间治疗后的第2 天对认知、姿势控制和潜在残留症状都得到了改善[43-44]。
综上所述,与传统抗失眠药物相比,daridorexant 起效机制不同,表现出良好的临床疗效和安全性,提高了患者的治疗依从性,展现出良好的临床应用前景。但daridorexant 上市时间较短,即便在获得批准之前开展了系列临床试验,其有效性与安全性仍需要在更广泛患者人群的临床应用中进一步评估。
猜你喜欢
中国心血管杂志(2022年2期)2022-11-25
中国心血管杂志(2022年4期)2022-11-25
高技术通讯(2021年3期)2021-06-09
中国心血管杂志(2021年6期)2021-01-02
科学(2020年5期)2020-11-26
特别健康·下半月(2019年6期)2019-08-01
祝您健康(2019年3期)2019-03-22
中国心血管杂志(2019年3期)2019-01-04
时代英语·高一(2018年5期)2018-11-19
时代英语·高一(2017年5期)2017-11-14
