
脓毒症相关肺损伤中内质网应激诱导的铁死亡机制研究
2024-04-17 09:35金子琦吴章宏
安徽医科大学学报 2024年3期
金子琦,唐 波,吴章宏,肖 宝,刘 斌,钟 扬,胡 霞
急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)是一种复杂的病理生理综合征,发病率高,但其发病机制仍有待探讨[1]。铁死亡是一种独特的铁依赖形式的程序性细胞死亡,其生化特征主要包括细胞内铁的积聚和大量脂质过氧化[2]。目前,铁死亡被认为是参与脂多糖(lipopolysaccharides,LPS)诱导的急性肺损伤(acute lung injury,ALI)的重要因素,用铁抑制素-1(ferrostatin-1,Fer-1)抑制铁死亡可显著缓解ALI[3]。因此,阻断铁死亡可能作为ARDS的一个新的治疗靶点。研究[4]表明,铁死亡受内质网应激(endoplasmic reticulum stress,ERs)的调节,如蛋白激酶RNA样ER激酶(protein kinase RNA-like-ER kinase,PERK)-激活转录因子 4(activating transcription factor 4,ATF4)途径参与介导铁死亡。在ERs情况下,内质网形状和功能通过内质网部分的选择性自噬得到良好维持[5]。内质网自噬由各种受体介导;其中,序列相似性家族134成员B(family with sequence similarity 134 member B,FAM134B)是在哺乳动物细胞中发现的第一个自噬体受体,其能靶向自噬体中的片段化内质网膜,用于溶酶体降解[6]。最近研究[7]显示FAM134B介导的内质网自噬保护脓毒症诱导的小鼠心肌损伤。此外,FAM134B介导的内质网自噬参与激活肝细胞癌铁死亡[8]。该研究通过探讨FAM134B介导的ER自噬和铁死亡之间的相互作用,旨在揭示ERs相关铁死亡在ARDS肺损伤中的作用机制。
1 材料与方法
1.1 材料
1.1.1细胞 小鼠肺泡上皮细胞(alveol epithelial cells,AECs)系MLE12细胞购自美国模式培养物集存库。
1.1.2实验动物 40只SPF级成年雄性C57BL/6J小鼠(6周龄,22~24 g)购自湖南斯莱克景达实验动物有限公司。所有小鼠饲养在湿度为50%~60%、温度为24~26 ℃且光照/黑暗周期为12 h的受控环境中。
1.1.3主要材料 CCK-8试剂盒(货号:CA1215-100T)、二甲苯(货号:DM0105)、苏木精和曙红(货号:G1120)、Triton X-100(货号:P1080)和二甲基亚砜(dimethyl sulfoxide,DMSO)(货号:D8370)购自北京Solarbio公司,脂多糖(lipopolysaccharide,LPS)(货号:L-2880)、脂质过氧化丙二醛(malondialdehyde,MDA)分析试剂盒(货号:MAK085)购自美国Sigma公司,铁分析试剂盒(货号:D153-6)、谷胱甘肽(glutathione,GSH)试剂盒(货号:A006-2)购自南京建成生物工程研究所,抗谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)(货号:CL594-67763)、β-肌动蛋白(货号:bsm-33036M)、环加氧酶2(prostaglandin endoperoxide synthase 2,PTGS2)(货号:66351-1-Ig)、葡萄糖调节蛋白78(glucose regulated protein 78,GRP78)(货号:66574-1-Ig)、ATF4(货号:60035-1-Ig)、CCAAT增强子结合蛋白同源蛋白(C/EBP homologous protein,CHOP)(货号:66741-1-Ig)、FAM134B(货号:21537-1-AP)、Alexa Fluor 568偶联的次级抗体(货号:A20185)购自美国Proteintech公司,DAPI(货号:H-1200)购自美国Vector Laboratory公司,组织蛋白提取物缓冲液(货号:89900)、BCA蛋白测定试剂盒(货号:23227)购自美国Thermo Fisher公司,4-羟基壬烯醛(4-HNE)(货号:GP2035)、山羊抗兔IgG二抗(货号:GB25303)、兔抗小鼠IgG二抗(货号:GB23303)购自武汉Servicebio公司。Vector、FAM134B过表达质粒(货号:D02008、M-21542)购自上海吉玛制药技术有限公司。
1.1.4主要仪器 DM2000荧光显微镜购自德国Leica公司,Tanon V8快速蛋白质印迹系统购自上海Tanon公司。
1.2 方法
1.2.1细胞分组与处理 MLE12细胞在37 ℃下在含有2%新生小牛血清的DMEM/F12培养基中培养。当细胞达到细胞培养瓶体积的80%~90%时,在6孔板中培养细胞。用LPS(0、1、2、5 μg/ml)处理细胞24 h以确定LPS对MLE12氧化应激和Fe2+水平的影响。为了验证铁死亡在LPS诱导的细胞死亡中的作用,将细胞分为对照(Con)组、铁死亡特异性抑制剂(Fer-1)组、LPS组和LPS+Fer-1组;LPS+Fer-1组在LPS处理前6 h,用10 μmol/L Fer-1预处理,随后将细胞暴露于5 μg/ml LPS (预实验确定的在24 h诱导几乎一半的MLE12细胞活力降低的浓度)24 h,Con组用融媒DMSO处理24 h,Fer-1组用10 μmol/L Fer-1预处理6 h,随后用DMSO处理24 h,LPS组将细胞暴露于5 μg/ml LPS 24 h;为了研究ER自噬在LPS引起的细胞铁死亡中的作用,将细胞分为Con+载体(Vector)组、Con+FAM134B组、LPS+Vector组、LPS+FAM134B组,细胞用Vector或FAM134B过表达质粒转染48 h后,暴露或不暴露于5 μg/ml LPS 24 h。
1.2.2CCK-8法测定细胞活力 采用CCK-8试剂盒说明书检测所有组MLE12细胞活力。
1.2.3铁含量测定 使用按照铁分析试剂盒说明书测量不同浓度(0、1、2、5 μg/ml)LPS处理组和Con组、Fer-1组、LPS组和LPS+Fer-1组细胞的铁浓度。将MLE12细胞(2×106个细胞)在6倍体积铁分析缓冲液中快速匀浆,加入相关试剂。最后,在593 nm处测量吸光度,并使用标准曲线计算铁浓度。
1.2.4实验动物分组和处理 将40只小鼠随机分成Con+Vector组、Con+FAM134B组、LPS+Vector组、LPS+FAM134B组,每组10只;LPS+Vector组、LPS+FAM134B组小鼠用1%戊巴比妥钠(40 mg/kg)进行麻醉,腹腔注射30 mg/kg LPS诱导脓毒症[9]。Con+Vector组、Con+FAM134B组小鼠腹腔注射相同剂量的PBS,Con+FAM134B组和LPS+FAM134B组小鼠在腹腔注射LPS前3天,用经鼻滴注的方法将基于腺相关病毒(adeno-associated virus,AAV)的FAM134B过表达质粒(1×1012vg/ml,60 μl)对小鼠进行干预,用于特异性表达FAM134B,腹腔注射LPS 12 h后处死小鼠。
1.2.5HE染色和免疫组化染色 将小鼠的整个左肺固定在4%多聚甲醛中,石蜡包埋,并切成4 μm的切片。然后将切片进行HE染色、4-羟基壬烯醛(4-Hydroxynonenal,4-HNE)免疫组化染色和GPX4免疫荧光分析。对于HE染色,将切片用苏木精和曙红染色,并固定在载玻片上,通过显微镜观察。对于免疫组化染色,组织切片首先在60 ℃下烘烤2 h,然后用二甲苯脱蜡,并通过乙醇梯度再水合。然后在枸橼酸盐缓冲液中于95 ℃加热30 min并冷却至室温,对载玻片进行抗原修复,阻断内源性过氧化物酶活性和血清封闭后,将切片与抗4-HNE单克隆抗体(1∶50)4 ℃孵育过夜,随后,将切片与生物素化连接的第二抗体在37 ℃孵育1 h,然后用DAB显色溶液进行DAB显色反应,细胞核复染和脱水后,用倒置显微镜拍摄组织切片图像。
1.2.6免疫荧光染色 切片用10%山羊血清和0.1% Triton X-100 37 ℃封闭1 h,然后加入抗GPX4(1∶100)初级抗体,4 ℃孵育过夜,然后将切片与Alexa Fluor 568偶联的次级抗体在37 ℃孵育1 h,用DAPI对细胞核进行染色。在荧光显微镜下观察并拍摄图像。
1.2.7Fe2+水平、MDA和GSH测定 收集肺组织(10 mg)或MLE12细胞(1×106个细胞),并使用Fe2+、脂质过氧化MDA分析试剂盒和GSH试剂盒测量MDA、GSH含量。
1.2.8蛋白质印迹法 采用组织蛋白提取物缓冲液制备小鼠肺组织及所有组的细胞的总蛋白,并使用BCA蛋白测定试剂盒进行定量。根据蛋白质分子大小使用10%或15% SDS-PAGE分离目标蛋白质,然后将蛋白质结合到PVDF膜上,随后用5%脱脂乳封闭3 h。此外,根据制造商提供的参考浓度,将PVDF膜与特异性一抗β-肌动蛋白(1∶2 000)、PTGS2(1∶800)、GPX4(1∶1 000)、GRP78(1∶1 200)、ATF4(1∶1 000)、CHOP(1∶1 000)和FAM134B(1∶1 000)在4 ℃孵育过夜,TBST洗涤3次后,PVDF膜与山羊抗兔或兔抗小鼠IgG二抗(1∶20 000)在室温下孵育2 h。最后,使用ECL蛋白印迹检测系统测定铁死亡标记物PTGS2和GPX4、ERs标志物GRP78、ATF4和CHOP以及介导内质网自噬的主要受体之一的FAM134B的蛋白水平。

2 结果
2.1 LPS对 MLE12细胞活力、铁死亡、氧化应激和Fe2+水平的影响CCK-8实验检测结果显示,LPS处理以剂量依赖性降低MLE12细胞活力(F=23.54,P<0.001),并且当LPS的浓度为5 μg/ml时,MLE12细胞活力下降至52%(图1)。LPS处理以剂量依赖性增加PTGS2和降低GPX4水平(F=21.77、28.72,均P<0.001)(图2),表明LPS暴露导致细胞铁死亡。此外,LPS处理还导致MLE12细胞中MDA、Fe2+的剂量依赖性增加(F=12.19、10.96,均P<0.001)(图3A、B),表明LPS暴露导致细胞内大量脂质过氧化和铁的积聚。此外,LPS处理以剂量依赖的方式降低了MLE12细胞中的GSH水平(F=18.64,P<0.001)(图3C),表明LPS处理使MLE12细胞抗氧化系统受损。

图1 不同浓度LPS对MLE12细胞活力的影响

图2 蛋白质印迹分析检测不同浓度LPS对MLE12细胞铁死亡的影响

图3 不同浓度LPS处理MLE12细胞中丙二醛、Fe2+含量和相对谷胱甘肽的比较
2.2 Fer-1减轻LPS诱导的MLE12细胞活力降低、铁死亡、氧化应激和Fe2+水平的增加如图4-6所示,与Con组相比,LPS组活力降低降低(t=4.28,P<0.001),PTGS2蛋白水平、MDA和Fe2+水平增加(t=12.03、5.52、8.17,均P<0.001),GPX4蛋白和GSH水平降低(t=4.60、4.98,P<0.001);与LPS组相比,LPS+Fer-1组细胞活力增加(t=3.51,P=0.005),PTGS2蛋白水平、MDA和Fe2+水平降低(t=6.90、3.07、3.50,均P<0.05),GPX4蛋白和GSH水平增加(t=3.39、3.45,P=0.008、0.006)。提示Fer-1能够减轻LPS诱导的MLE12细胞活力降低、铁死亡、氧化应激和Fe2+水平的增加。

图4 Fer-1对LPS诱导的MLE12细胞活力降低的影响
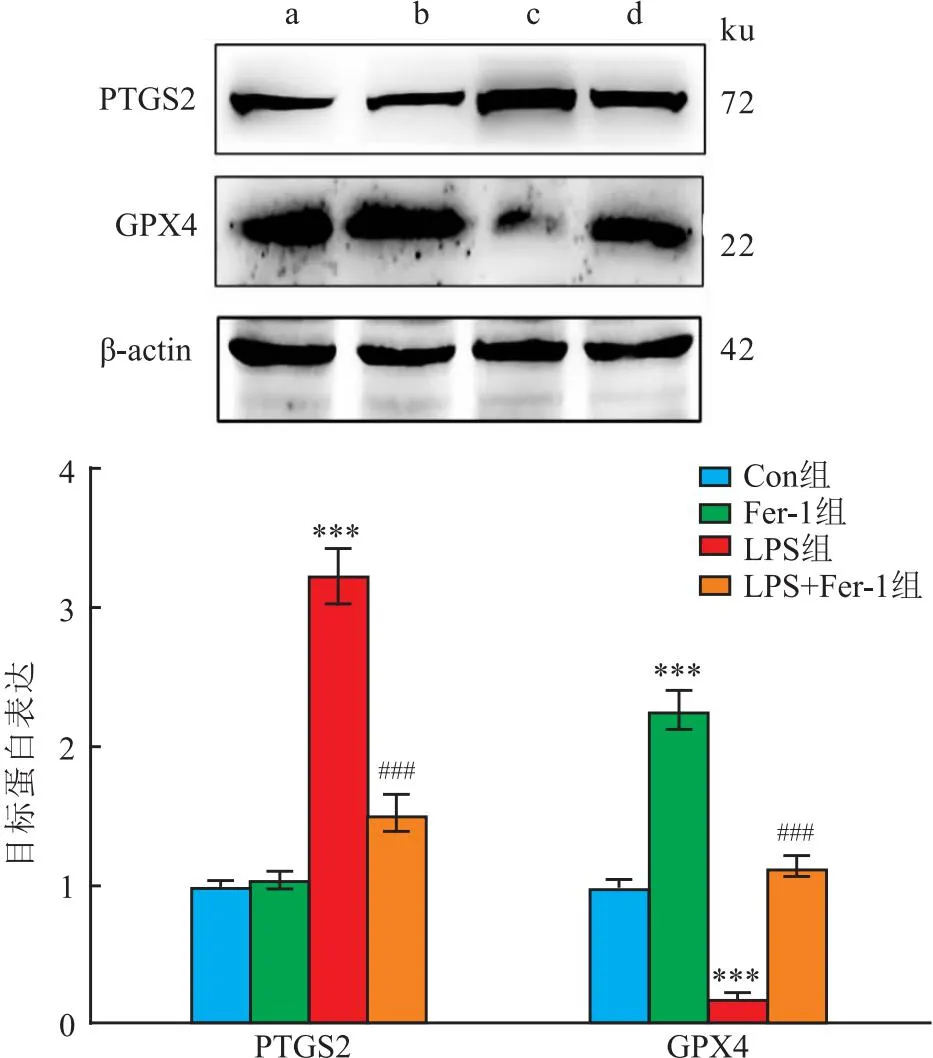
图5 Fer-1对LPS诱导的MLE12细胞铁死亡的影响
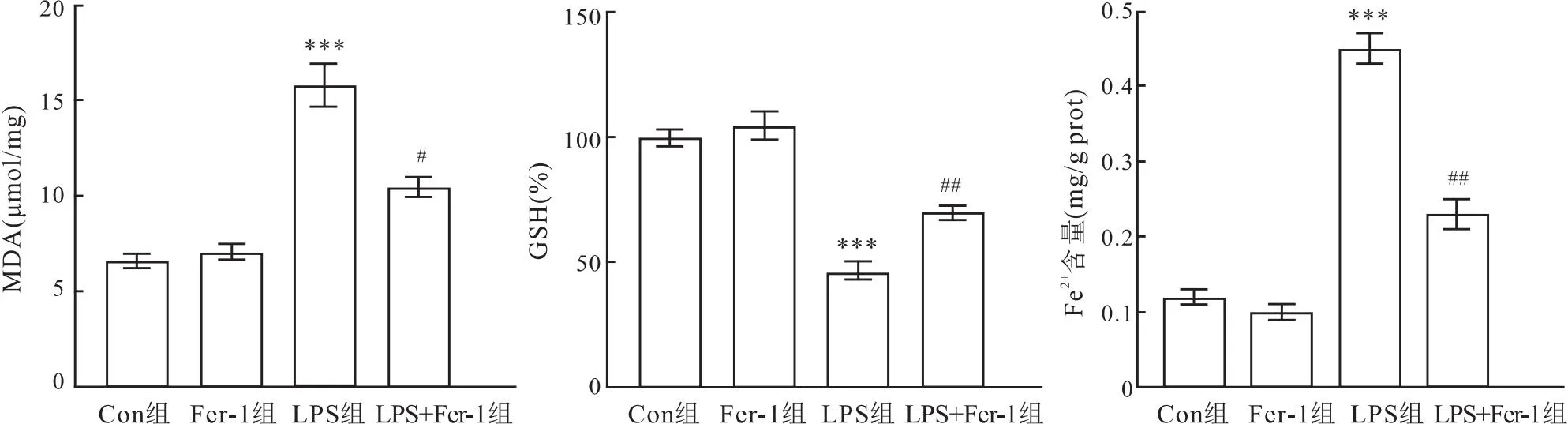
图6 Fer-1对LPS诱导的MLE12细胞氧化应激和Fe2+水平增加的影响
2.3 FAM134B通过抑制ERs改善LPS诱导的细胞活力降低、铁死亡和氧化应激增加LPS处理以剂量依赖性增加MLE12细胞中GRP78、ATF4和CHOP水平(F=11.63、24.92、10.10,均P<0.001),降低FAM134B水平(F=19.72,P<0.001)(图7)。表明LPS诱导MLE12细胞ERs并抑制内质网自噬。与LPS+Vector组相比,LPS+FAM134B组细胞中ATF4、CHOP和PTGS2蛋白水平降低(t=6.50、5.83、5.38,P<0.001),GPX4和FAM134B蛋白水平增加(t=6.22、6.65,P<0.001),细胞活力增加(t=3.526,P=0.004)(图8-10)。同时,LPS+FAM134B组细胞的MDA水平低于LPS+Vector组(t=3.11,P=0.021),和GSH水平高于LPS+Vector组(t=3.41,P=0.006)(图11)。提示FAM134B通过抑制ERs改善LPS诱导的细胞活力降低、铁死亡和氧化应激增加。
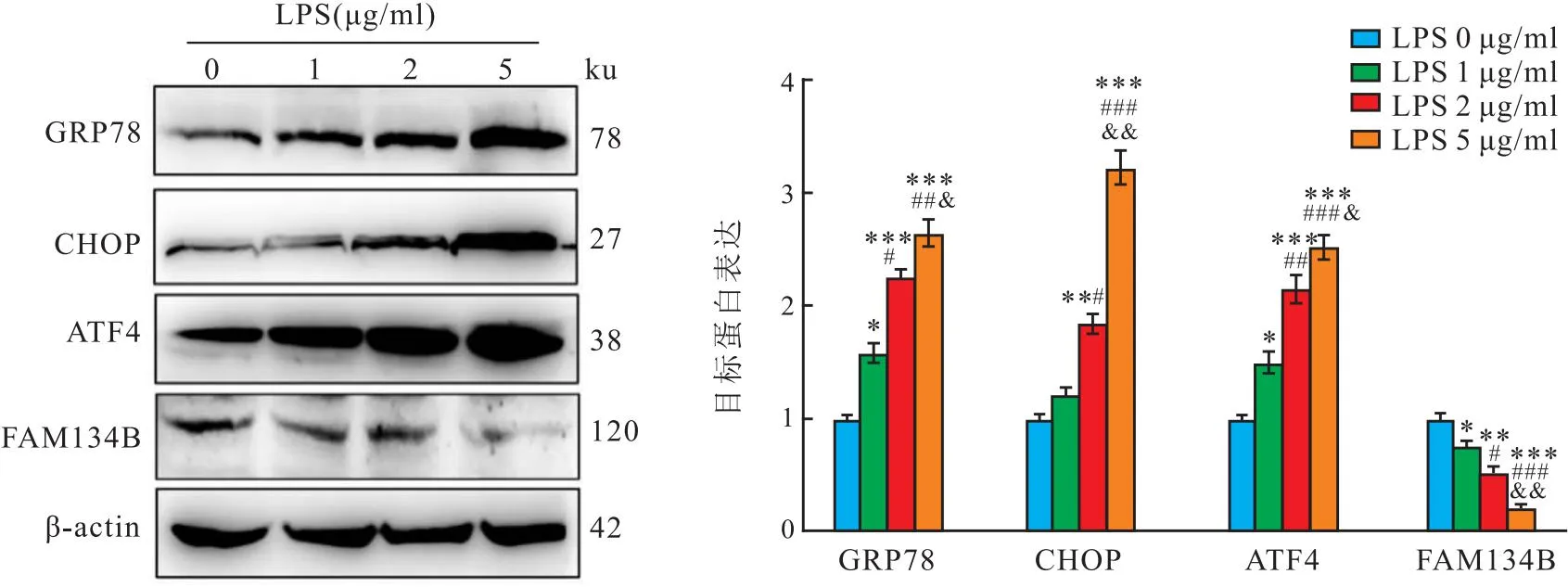
图7 不同浓度LPS对MLE12细胞ERs和内质网自噬的影响
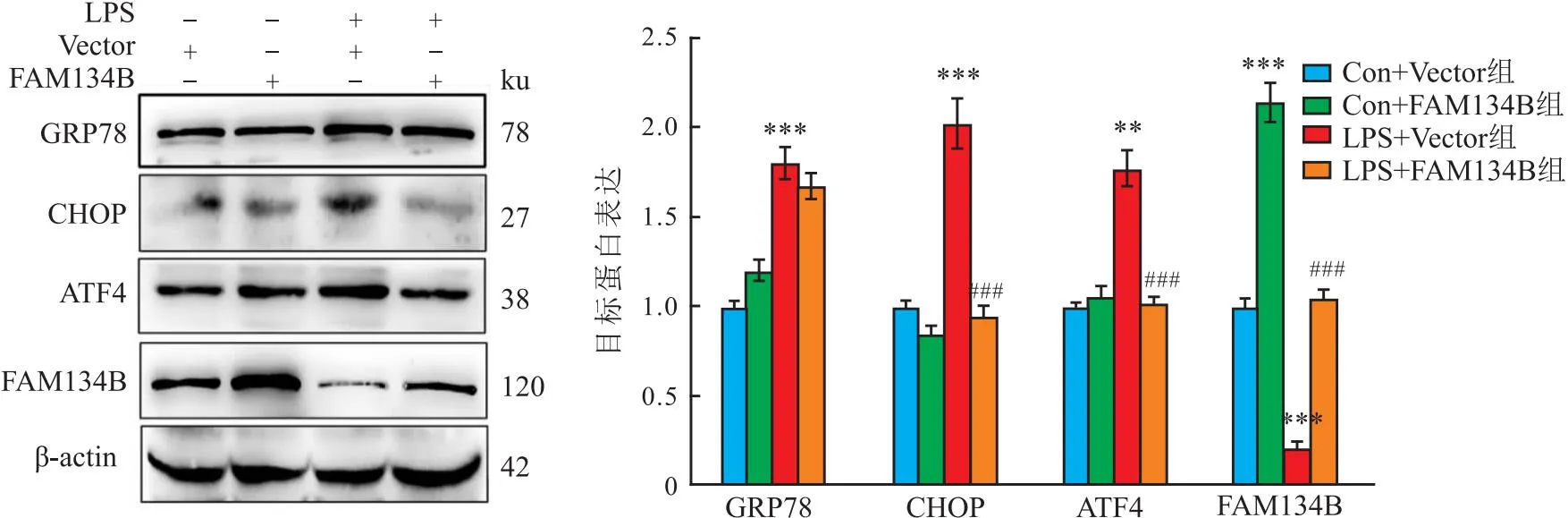
图8 FAM134B对LPS诱导的MLE12细胞ERs和内质网自噬抑制的影响
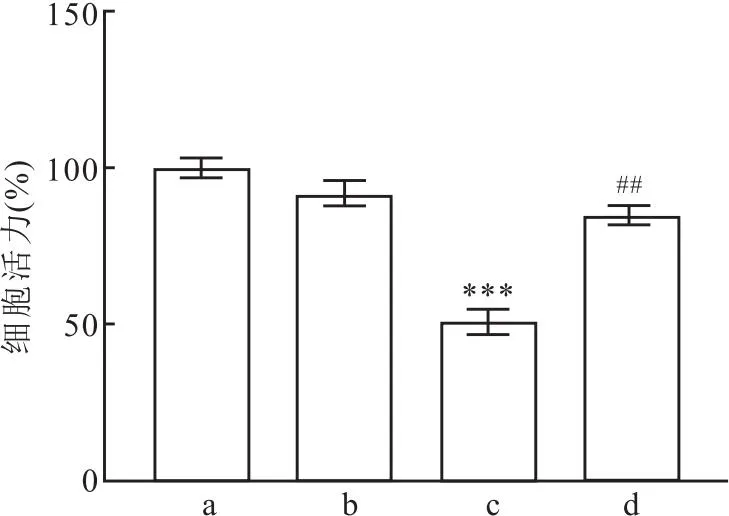
图9 FAM134B对LPS诱导的MLE12细胞活力降低的影响
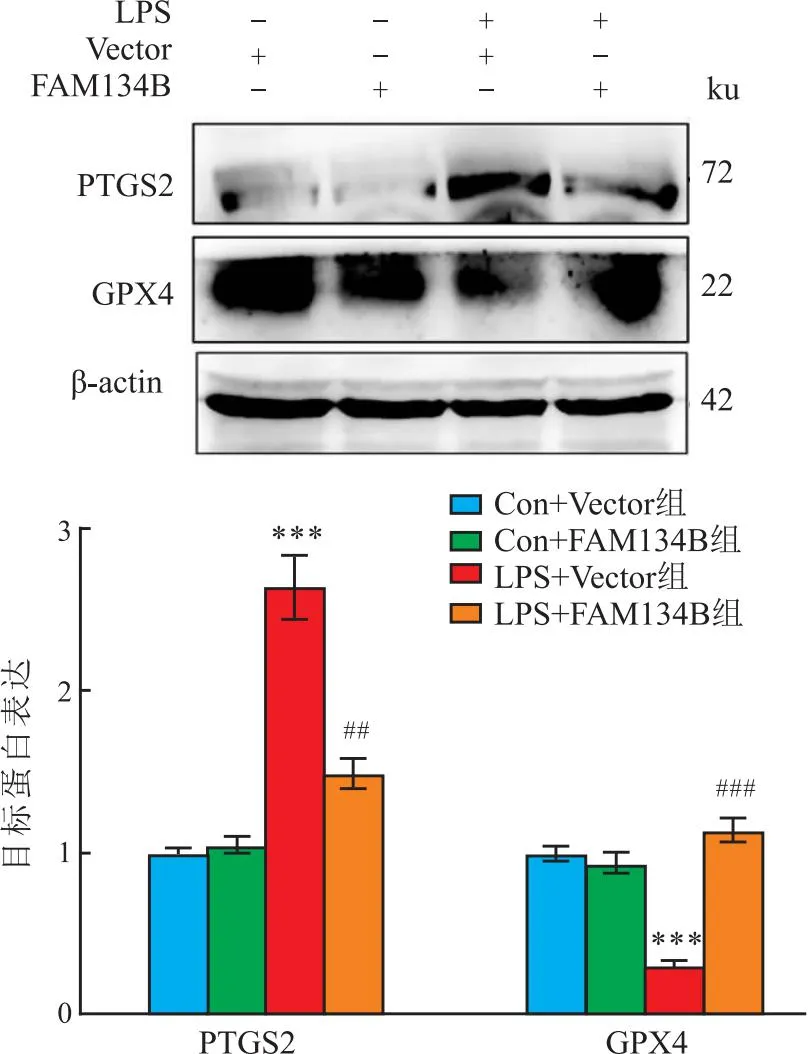
图10 FAM134B对LPS诱导的MLE12细胞铁死亡的影响
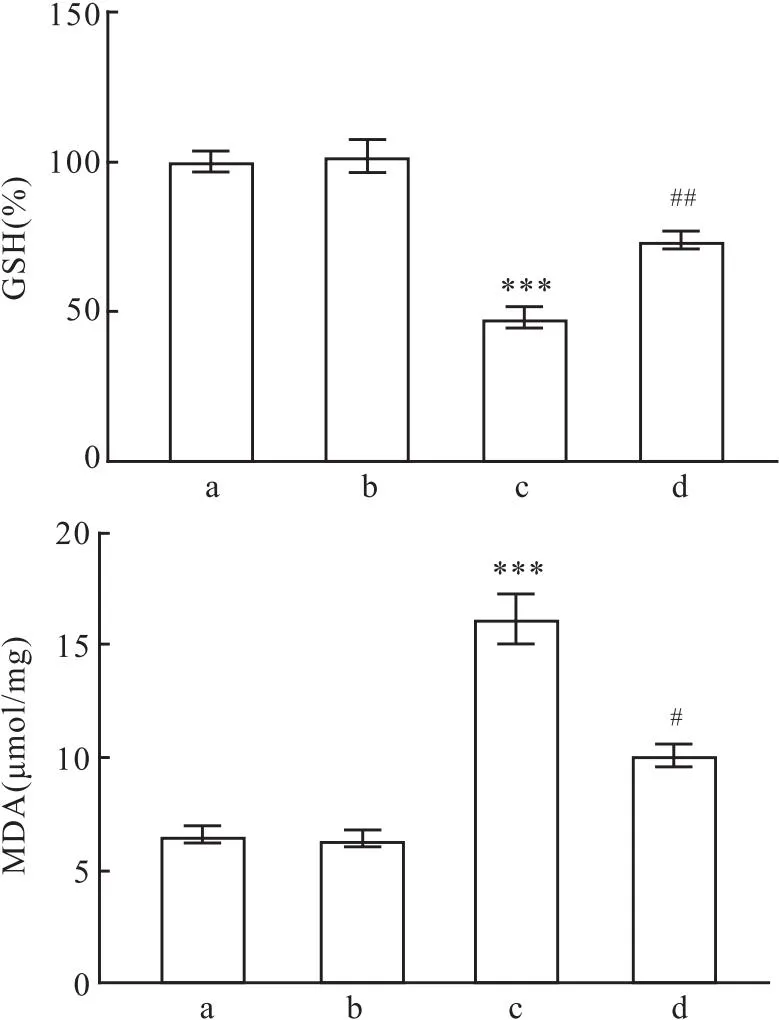
图11 FAM134B对LPS诱导的MLE12细胞氧化应激的影响
2.4 FAM134B过度表达减轻LPS诱导的小鼠肺部炎症损伤HE染色分析结果显示,与Con+Vector组小鼠相比,LPS+Vector组小鼠肺组织中出现中性粒细胞浸润、肺泡隔膜增厚、肺泡水肿。FAM134B+LPS组小鼠肺组织中中性粒细胞浸润、肺泡隔膜增厚以及肺泡水肿较LPS+Vector组减轻(图12A);与Con+Vector组小鼠相比,LPS+Vector组小鼠肺组织中4-HNE、ATF4和CHOP表达水平增加(t=5.36、3.87、4.35,均P<0.01),GPX4、FAM134B表达水平降低(t=5.07、5.64,均P<0.001)。说明LPS诱导小鼠肺组织ERs并抑制内质网自噬。与LPS+Vector组相比,LPS+FAM134B组小鼠肺组织中4-HNE、ATF4、CHOP表达水平降低(t=3.88、3.55、5.61,均P<0.01),GPX4、FAM134B表达水平增加(t=2.87、5.07,均P<0.05)(图12B-G)。说明FAM134B过度表达减轻LPS诱导的小鼠肺部炎症损伤。
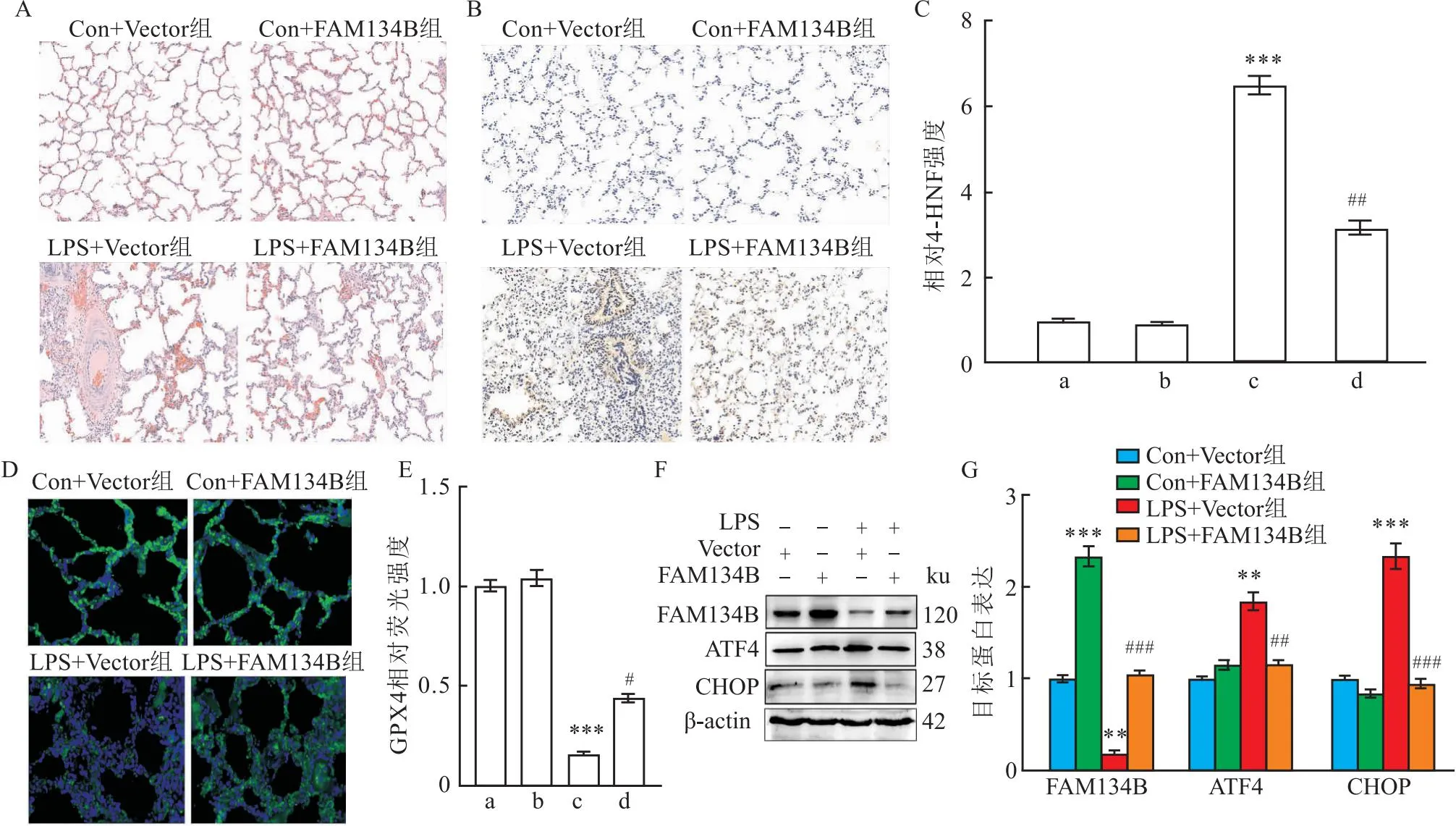
图12 FAM134B过度表达对LPS诱导的小鼠肺部损伤的影响(n=6)
3 讨论
铁死亡是最近发现的一种氧化性细胞死亡,其特征是铁依赖性脂质过氧化,并与多种疾病过程有关,包括癌症、缺血再灌注损伤、感染和神经退行性疾病[5]。肺中的各种细胞类型,包括上皮细胞和巨噬细胞,可以产生铁代谢相关蛋白,以调节铁稳态,保护肺组织免受氧化应激[2]。铁代谢紊乱与ARDS患者的肺组织损伤密切相关[3],即过多的铁可以通过芬顿反应产生活性氧和细胞毒性。临床研究[10]表明,ARDS的严重程度与铁和铁相关蛋白的水平有关。研究[11]表明,血液制品中的铁会导致受血者体内铁的增加,从而促进输血相关ALI的发生。在ARDS患者的支气管肺泡灌洗液中可以检测到Fe2+和铁调节剂水平升高[12]。由于肺泡上皮细胞死亡可能是ARDS中肺泡损伤的一个关键特征,受损的AECs产生过量活性氧进一步加重ARDS肺损伤[13]。因此,本研究选择MLE12细胞作为体外研究对象。先前研究[14]表明Fer-1抑制脂质过氧化减轻了LPS诱导的人支气管上皮细胞系BEAS-2B细胞死亡。本研究发现LPS以剂量依赖的方式诱导MLE12细胞活力降低、铁死亡、氧化应激增加,这与先前的研究一致。
众所周知,内质网是一种重要的细胞器,对活性氧敏感。活性氧在内质网中的持续积聚引发ERs,可能是导致ALI进展的重要机制[4]。证据[3]表明,抑制ERs可以改善脓毒症相关的肺损伤。GRP78是ERs的主要调节蛋白,ATF4在ERs中显著上调,促进CHOP等因子表达,这些因子通常被视为ERs的生物标志物[3]。本研究发现,随着LPS剂量的增加,GRP78、ATF4和CHOP的表达水平增加,表明LPS可以诱导MLE12细胞的ERs。近年来,证据[7]表明ERs增加与LPS诱导的铁死亡相关。FAM134B作为介导内质网自噬的主要受体之一,在ERs情况下,可以通过螯合进入自噬体来介导ER进入溶酶体。FAM134B可以通过其膜弯曲能力促进膜重塑和减轻ERs破坏,并通过与LC3的结合诱导内质网自噬,FAM134B的下调将导致内质网的扩张[7]。此外,研究[15]表明,未折叠蛋白反应(unfolded protein response,UPR)可以激活FAM134B的表达以识别内质网腔中错误折叠或聚集的蛋白质。本研究发现LPS激活了MLE12细胞中的ERs和抑制内质网自噬,通过上调FAM134B,减轻了LPS诱导的MLE12细胞活力降低、铁死亡和氧化应激增加,提示内质网自噬抑制参与介导LPS诱导的MLE12细胞铁死亡。此外,本研究通过体内研究验证了体外发现,LPS诱导的小鼠肺部组织中4-HNE上调和GPX4表达抑制,伴随着ERs激活和FAM134B水平的下降,而FAM134B预处理逆转了LPS诱导的这些变化。研究[7]表明,脓毒症相关肺损伤中4-HNE超载通过上调ERs基因(如ATF4、CHOP)激活所有UPR途径,并促进铁死亡。因此,上调FAM134B对于抑制ARDS病理过程相关的铁死亡至关重要。
总之,本研究显示LPS以剂量依赖的方式诱导MLE12细胞铁死亡和ERs。通过激活内质网自噬相关FAM134B受体有助于抑制ERs,并减轻细胞铁死亡。本研究的发现为LPS诱导的ARDS提供了新的见解,这有助于更好地理解ARDS相关病理机制,并作为寻找预防ARDS的有效策略的基础。然而,本研究主要集中在FAM134B自噬受体上,不能排除其他受体以及其他形式的自噬(如铁蛋白自噬)是否参与LPS诱导的肺上皮细胞铁死亡,未来需要更多研究以扩展本研究发现。
猜你喜欢
解放军医学杂志(2021年12期)2022-01-18
服饰导报·鞋世界(2021年4期)2021-05-17
现代临床医学(2021年1期)2021-01-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国药理学通报(2019年5期)2019-01-11
安徽医科大学学报(2016年12期)2017-01-15
西南军医(2016年6期)2016-01-23
中国当代医药(2015年33期)2015-03-01
西南军医(2015年2期)2015-01-22
食品科学(2013年15期)2013-03-11
