
强直性肌营养不良一家系临床分析
2012-09-30 06:39张振铎
中国实用神经疾病杂志 2012年12期
张振铎
郑州大学一附院在职博士(河南中医学院第一附属医院神经内科)郑州 450000
强直性营养不良 (myotonic dystrophy,DM),是一种常染色体显性遗传病。临床以肌强直、肌无力和肌萎缩为主要特征,伴有内分泌异常、视力减退、心脏受累等多系统损害。起病隐匿、进展缓慢,临床表现复杂多样。本研究对一临床家系进行分析,探讨该病的临床特点,旨在提高对该病的认识水平。
1 临床资料
5例患者为同一DM家系成员,来自我院神经内科门诊。该家系谱如图1所示。
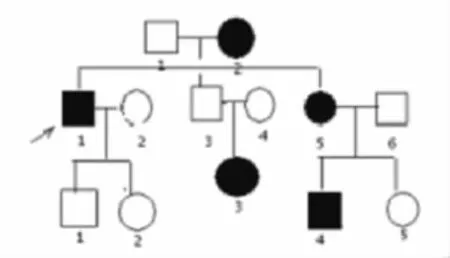
图1 DM一家系谱

图2 先证者左肱二头肌活检骨骼肌纤维大小不等,可见肌核内移,肌浆块和镶边空泡(改良Gomori染色×300)
该家系成员共13人,DM 患者5人(男2人,女3人)。先证者男,38岁,5a前出现双手握力减弱,用力握拳后放松困难,反复活动后症状可减轻,久坐后起身困难,行走呈鸭步,蹲下站起、负重行走费力,并感视力减退。病情呈进行性加重且出现脱发、性功能下降。无肢体麻木、疼痛、肌肉跳动等症。体格检查:前额秃顶,斧状脸,心率80次/min,律齐,各瓣膜听诊区未闻及病理性杂音。神经系统:神清,记忆力减退。双眼白内障,左眼视力0.8,右眼0.6,张口费力,咀嚼、伸舌动作僵硬。双侧胸锁乳突肌、鱼际肌、肱二头肌、股四头肌萎缩,肱二头肌叩击可见肌球形成,持续约2min消失。双上肢近端肌力Ⅲ级,远端肌力Ⅳ级,双下肢近端肌力Ⅳ级弱,远端肌力Ⅳ级,双侧腱反射减弱,肱二头肌、腓肠肌叩诊可见肌球形成,指间肌群轻度萎缩,双侧病理征阴性。辅助检查:肌酸激酶(CK)438U/L,乳酸脱氢酶(LDH)322U/L;双眼裂隙灯检查:双侧晶体后囊中心白色浑浊,考虑白内障心电图检查:窦性心率,左束支传导阻滞。肌电图(EMG):胸锁乳突肌、肱二头肌、股四头肌运动单位动作电位波幅降低、时限缩短,符合肌源性损害表现;放松时可见肌强直电位发放。左肱二头肌活检:横纹肌萎缩,肌纤维大小不等,肌核内移,可见肌浆块和镶边空泡(见图2)。其余4例DM患者的临床资料见表1。

表1 4例DM患者的临床资料
2 讨论
DM是一种常染色体显性遗传病[1],与DM 相关的基因有2型:DM1型和 DM2型。DM1型[2]位于19q 13.2-13.3,编码强直性肌营养不良蛋白激酶(DMPK),由于该基因3’端非翻译区内三核苷酸(CTG)n重复序列拷贝数增加而发病。正常人(CTG)n重复序列一般小于37。等位基因的长度大于37个CTG重复序列通常是不稳定的,在有丝分裂过程中长度可能会增加,下一代通常会遗传到更长的重复序列,从而产生遗传早现现象,即子代较父代发病年龄更低,病情更重,以致出现先天性DM1[3]。具有38~49个(CTG)n重复序列为突变临界状态,携带者可能表现正常,而下一代会有更高的重复率及发病可能。(CTG)n重复序列的长度大于50被认为是完全突变,且重复序列的长度与疾病的严重程度及发病年龄密切相关,即重复序列越长,症状越重,发病年龄越小[4]。DM2型[5]位于3q 13.3~24,编码锌指蛋白9(ZNF9),由其1号内含子中的表达核苷酸(CCTG)n重复序列拷贝数增加而发病。
事实上,由于重复的扩增序列位于DMPK和ZNF9非翻译编码区域,并不直接改变所在基因区域的蛋白编码[6]:在DM1扩增的CTG重复序列转录表达为扩增的CUG-RNA重复序列 (expanded CUG repeat RNA,CUGexp);在DM2扩增的CCTG转录表达为扩增的CCUG-RNA重复序列(expanded CCUG repeat RNA,CCUG-RNAexp)。研究发现这两种扩增的RNA重复序列在受损的组织的细胞核内聚集,形成境界清楚的核糖核包涵体(ribonuclear inclusions)。核糖核包涵体在核内的聚集引起 MBNL-1(muscle blind-like 1)和CUGBP-1(CUG-blinding protein 1)功能失调。而 MBNL-1和CUGBP-1是调节细胞内pre-mRNA选择性剪接的相互拮抗的调节因子,选择性剪切的调节错误引起某些基因如肌钙蛋白、胰岛素受体及肌肉中氯离子通道等编码的基因在表达过程中发生拼接错误而产生相应的临床表现。
DM主要临床特征是以骨骼肌受损为主的多系统受累肌病[7]。骨骼肌受损的临床主要表现为肌无力、肌萎缩、肌强直。DM1早期以远端肌肉受累为主,上肢远端受累引起手的精细动作不能,下肢远端受累引起足下垂和踝背曲力弱。亦常累及头面部肌肉,甚至可以累及呼吸肌。头面部颞肌、咀嚼肌萎缩,颧骨突出,面容瘦长,呈典型斧状脸,成为本病特征性面容。胸锁乳突肌萎缩致颈部细长,伸颈肌力减弱伴过度前伸,呈“鹅颈”。肌强直现象表现为肌肉起动困难,收缩后不能放松,常影响患者的日常功能,如使用工具、旋转门把手等。肌强直具有热生现象(warm up phenomenon)[8],即反复活动后强直症状减轻。典型的肌强直易通过叩击鱼际肌或舌肌所引出,形成肌球征,被称为叩击性肌强直(percussion myotonia)。DM2早期多累及肢体近端肌肉,如肘伸肌、臀屈肌等,较少累及头面部肌肉、呼吸肌。心肌受累主要表现为心房纤颤、窦性心动过缓、房室传导阻滞等心律失常和心肌病[9],部分患者可能因房室传导阻滞而猝死[10]。白内障为本病的主要眼部症状甚至首发症状,一部分患者甚至在没有出现骨骼肌系统表现时即出现晶状体受累而出现视力减退[11]。Kim等[12]报道,除白内障外,视网膜变性也是DM患者视力减退的原因之一,眼底检查可见视网膜周边色素上皮细胞褪色。性腺内分泌系统主要由于性腺发育不良或原发性腺功能衰竭,男性表现为睾丸萎缩、阳痿、性功能减退等,女性表现为卵巢萎缩或卵巢功能不全、闭经或月经失调、不孕或习惯性流产等。胰岛素对糖负荷反应增高,出现糖耐量异常,但糖尿病的发病率并不高[13]。神经系统主要表现为认知功能下降,大脑白质受损,引起皮质间联系中断可能是其认知功能下降原因之一[14]。
DM实验室检查肌酶轻中度升高。肌电图呈肌强直电位及运动时限缩短,波幅减低,重收缩时出现病理性干扰项等典型肌源性改变[15]。肌肉活检典型骨骼肌病理改变为肌纤维大小不等,相嵌分布,肌膜核增多、核内移,排列呈链状,肌原纤维向一侧退缩形成肌浆块,Ⅰ型纤维萎缩、Ⅱ型纤维肥大,核内移早期出现在萎缩的Ⅰ型纤维内,随着肌力的减弱,核内移明显增加,到晚期,大多数萎缩肌纤维和肥大肌纤维含有一个或多个内移的核[16]。
本组5例患者均有不同程度的肌强直、肌无力,其中4例有肌萎缩、1例肌萎缩不明显考虑与病程较短有关。患者自觉肢体僵硬,握拳放松困难、静坐后迈步困难、用力咀嚼后松口困难等,重复动作后症状减轻。叩击舌肌、大鱼际肌、前臂或腿部肌群可有肌球形成。实验室检查血清肌酶(CPK、LDH等)轻度升高或正常。肌强直放电和肌源性损害是本病的重要EMG特征,本组肌强直放电出现于全部患者,说明EMG可作为本病临床诊断的主要辅助检查方法。本组患者肌肉活检,其病理学改变与以往的研究报道相同,主要表现为不同程度的肌纤维萎缩、核内移、肌浆块等,符合DM的诊断标准[15]。从临床特点看,该家系患者主要表现为肢体远端肌肉受累,呼吸肌无明显受累,符合DM2型,类型确诊有待于基因检测。
目前该病尚无有效的治疗方法,主要是对症处理,如服用卡马西平、丙戊酸钠控制肌强直,晶体植入治疗白内障,安装起搏器治疗心律失常等。针对不同靶点的基因治疗如抑制重复序列的扩增、修复突变的DMPK-RNA、改变RNA结合蛋白水平等具有一定的前景[17]。该家系2例患者经中医辨证施治1a来病情稳定,机制有待进一步探讨。
[1]Mulders S,van Engelen B,Wieringa B,et al.Molecular therapy in myotonic dystrophy:focus on RNA gain-of-function[J].Human Molecular Genetics,2010,19:90-97.
[2]Brook JD,McCurrach ME,Harley HG,et al.Molecular basis of myotonic dystrophy:expansion of a trinucleotide(CTG)repeat at the 3-prime end of a transcript encoding aprotein kinase family member[J].Cell,1992,68:799-808.
[3]Rakocevic-Stojanovic V,Savic D,Pavlovic S,et al.Intergenerati onal changes of CTG repeat depending on the sex of the transmitting parent in myotonic dystrophy type 1[J].Eur J Neurol,2005,12:236-237.
[4]Erin PF,Mani S,Mahadevan.Therapeutics development in mytonic dystrophy type 1[J].Muscle Nerve,2011,44:160-169.
[5]Liquori CL,Ricker K,Moseley ML,et al.Myotonic dystrophy type 2caused by a CCTG expansion in intron 1of ZNF9[J].Science,2001,293:864-867.
[6]Wheeler TM.Myotonic dystrophy:Therapeutic strategies for the future[J].Neurotherapeutics,2008,5:592-600.
[7]Chris T,David HJ.The myotonic dystrophies:diagnosis and management[J].J Neurol Neurosurg Psychiatry,2010,81:358-367.
[8]Logigian EL,Blood CL,Dilek N,et al.Quantitative analysis of the"warm-up"phenomenon in myotonic dystrophy type 1[J].Muscle Nerve,2005,32:35-42.
[9]Alide A,Tieleman KM,Jenks JS,et al.High disease impact of myotonic dystrophy type 2on physical and mental functioning[J].J Neurol,2011,258:1 820-1 826.
[10]Jorge P,Emilce T,Mauricio A,et al.Progressive conduction disturbancein myotonic dystrophy[J].Cardiology Journal,2011,18(3):322-325.
[11]Garrott HM,Walland MJ,O’Day J.Recurrent posterior capsular opacification and capsulorhexis contracture after cataract surgery in myotonic dystrophy[J].Clin Experiment Ophthalmol,2004,32:653-655.
[12]Kim US,Kim JS,Hwang JM,et al.A case of myotonic dystrophy with pigmentary retinal changes[J].Korean J Ophthalmol,2009,23(2):121-123.
[13]Gaul C,Schmidt T,Windisch G,et al.Subtle cognitive dysfunction in adult onset myotonic dystrophy type 1(DM1)and type 2(DM2)[J].Neurology,2006,67:350-352.
[14]Jeffrey RW,Bryon AM,Erin E,et al.White matter abnormalities and neurocognitive correlates in children and adolescents with myotonic dystrophy type 1:A diffusion tensor imaging study[J].Neuromuscul Disord,2011,21(2):89-96.
[15]王新德 .神经病学[M].北京:人民军医出版社,2007:282-287.
[16]吴江 .神经病学[M].北京:人民卫生出版社,2007:355-356.
[17]Magan JJ,Bulmaro C.Perspectives on gene therapy in myotonic dystrophy type 1[J].Journal of Neuroscience Research,2011,89:275-285.
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
保健与生活(2022年13期)2022-07-06
临床骨科杂志(2021年6期)2022-01-08
疯狂英语·新阅版(2021年8期)2021-09-10
国际放射医学核医学杂志(2021年10期)2021-02-28
北京广播电视报(2019年8期)2019-03-27
武警医学(2019年2期)2019-03-05
中国医学科学院学报(2019年1期)2019-03-05
食品安全导刊(2018年36期)2018-05-25
