
青霉素和阿莫西林的电子结构及化学稳定性
2012-12-25 02:08张宏森杨学舜解丽萍
黑龙江科技大学学报 2012年1期
张宏森, 杨学舜, 张 钢, 解丽萍
(1.黑龙江科技学院 现代分析测试研究中心,哈尔滨150027;2.哈尔滨工程大学 材料科学与化学工程学院,哈尔滨150001;3.吉林大学理论化学研究所 理论化学计算国家重点实验室,长春130023)
青霉素和阿莫西林的电子结构及化学稳定性
张宏森1,2, 杨学舜1, 张 钢3, 解丽萍1
(1.黑龙江科技学院 现代分析测试研究中心,哈尔滨150027;2.哈尔滨工程大学 材料科学与化学工程学院,哈尔滨150001;3.吉林大学理论化学研究所 理论化学计算国家重点实验室,长春130023)
青霉素类抗生素化学稳定性问题一直是理论研究和医药生产的热点和难点。选取青霉素、阿莫西林两种代表性药物,采用精确的杂化密度泛函理论(B3LYP)方法,从分子几何构型、键能、价键轨道、静电势和化学硬度等方面分析了化学结构对于化学稳定性的影响。结果表明:稠环张力是导致该类化合物不稳定的主要原因,取代基影响分子的电子排布和分子间的相互作用。该理论研究为青霉素类抗生素新药设计、构效关系和化学反应规律的研究,以及医药生产中产品质量的控制提供了理论参考。
阿莫西林;青霉素;密度泛函理论;化学稳定性
0 引言
青霉素自从1941年问世以来,青霉素类抗生素以其高效低毒、选择性强、抗菌谱广、体内分布好等特点,挽救了数以千百万计的生命,至今依旧是举世公认的重要的临床抗感染药物。然而研究表明:青霉素类化合物化学性质不稳定,易发生分解、聚合等副反应。这不但对生产、储运提出了更高的要求,而且副反应产物严重地影响产品质量,甚至威胁到用药安全。例如,各种青霉素副产物的衍生物是导致“青霉素过敏”反应的重要原因[1]。尽管科研人员进行了大量的卓有成效的研究工作,但依旧难以从本质上解决青霉素类抗生素稳定性问题。
众所周知,“结构决定性质”,开展该类化合物电子结构的基础研究有利于从本质上认识稳定性等相关性质。自从1949年霍奇金女士第一次成功测定了青霉素的结构起,人类就从未间断过对青霉素类抗生素构效关系的探索。近年来,随着理论化学取得的长足进步,特别是密度泛函理论在化学领域的广泛应用,对于以青霉素为代表的β-内酰胺类化合物研究进入了崭新的时代。大量研究表明:密度泛函理论是研究β-内酰胺类化合物结构和性质的重要理论工具[2-4]。
在众多的β-内酰胺类化合物中,青霉素和阿莫西林是该类药物中最为常见的两种药物,二者结构相近,但是它们在化学性质方面却表现出一定的差异。例如:在酸性条件下,阿莫西林的稳定性明显优于青霉素,因此更适合制成口服制剂。应用密度泛函理论对这两种代表性药物进行研究,有利于分析该类化合物所具有的共同性质,以及相应取代基对于分子整体化学性质的影响,为进一步的相关研究奠定理论基础。
1 实验与方法
采用包括电子交换相关效应的杂化密度泛函理论(B3LYP)方法,结合6-31G(d)基组,优化分子。振动分析表明,所有的优化结构均没有虚频,都是势能面上的局部极小点。以优化后的结构为基础分析了分子的前线轨道能级、静电势图和化学硬度。采用了NBO(nature bond orbital)的方法分析了其电荷分布以及轨道之间和孤对电子与轨道之间的相互作用。应用极化连续介质模型(Polarized Continuum Model)模拟溶剂化效应。文中所有计算均使用Gaussian 03 程序完成[5]。
2 结果与讨论
2.1 几何结构
笔者对阿莫西林和青霉素的分子结构进行了几何优化。图1为阿莫西林和青霉素的分子结构,表1给出了优化后的几何参数以及阿莫西林和青霉素对应的实验值。

表1 在6-31G(d)水平下青霉素、阿莫西林分子的主要几何参数以及相应实验值Table 1 Domain geometry parameters of penicillin and amoxicillin molecule optimized at 6-31G(d)and experimental level
由图1可知,阿莫西林和青霉素的母核都是由β-内酰胺环和氢化噻唑环骈合而成,由二面角C4—N1—C2—S7和 C5—N1—C2—C3可知,两个稠环并不共面。β-内酰胺环是该类化合所必须的生物活性基团。其中,由于氮原子与碳原子在半径和电负性方面的差异,导致四元环对边C—C和C—N键的键长不对称,二面角 C4—N1—C2—C3为-13.529 5°,说明四元环结构发生扭曲,张力增大。这是造成四元环结构不稳定容易开环的主要原因。同理,C—S与C—C键长的差异导致了氢化噻唑环张力的增大,使其易于开环断裂,结构不稳定。
从表1的数据可以看出,阿莫西林和青霉素四元环中 N1—C4键键长分别处于典型的 N==C(0.128 7 nm)双键和N—C(0.147 2 nm)单键之间,说明N1与C4==O13之间发生共轭形成π键。但其对电子和C4==O13如果存在共轭作用,会降低N原子受到亲电试剂的进攻能力,增加结构的稳定性。
2.2 键能
键能作为表征化学键的重要参数,被广泛用于研究化合物的化学反应活性及其热力学性质,尤其在化学稳定性研究方面具有重要价值。然而,目前实验难以获得的化学键逐级解离能。研究表明:采用密度泛函理论计算方法可以给出合理的理论预测值[8],对于化学键A—B,其键解离能(Bond Dissociation Energy,BDE)定义为化学键对应解离通道的焓变:键长更加接近于N—C单键,说明共轭作用并不明显。通常认为β-内酰胺环上N原子中的孤对电子,易受到亲电试剂的进攻,导致开环,而N1上的孤

表2显示,青霉素和阿莫西林稠环内的化学键的键能基本一致。这说明相应取代基的差异并没有影响到环中化学键的键能。环内C2—C3、C3—C4键能明显小于环外C5—C8键能,这再次表明该类化合物的主要不稳定部位应为β-内酰胺环。C6—S7键能小于C2—S7键能,而且能量较低,因此C6—S7应优先断裂。这与前期研究中的实验检测结果相吻合[9]。
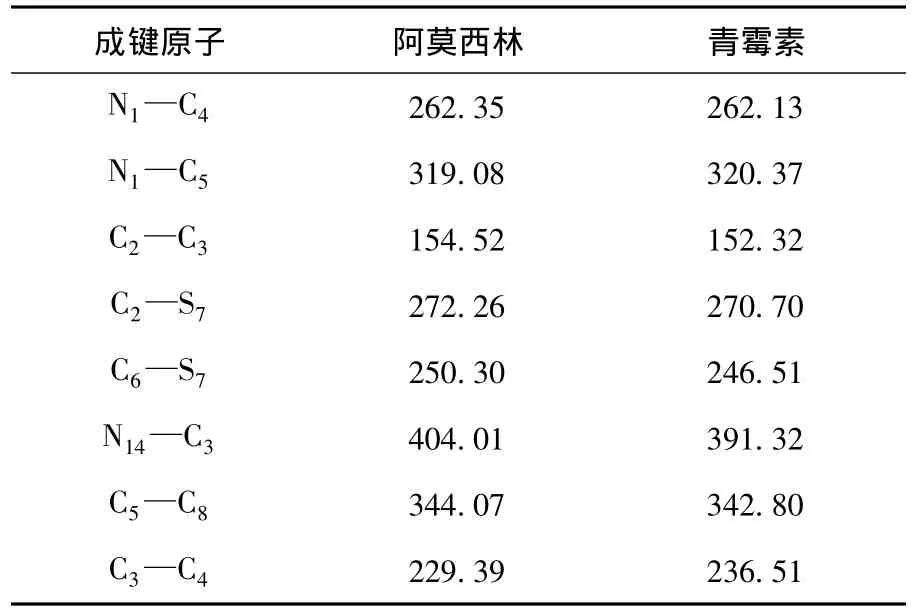
表2 6-31G(d)水平下计算青霉素、阿莫西林分子部分键解离能Table 2 Predicted domain bond dissociation energy of penicillin and amoxicillin at B3LYP/6-31G(d)level kJ/mol
2.3 自然键轨道
自然键轨道分析可以详细地分析成键情况和键—键相互作用。表3列出了阿莫西林和青霉素电子供体(Donor)轨道i、电子受体(Acceptor)轨道j以及由二级微扰理论得到的它们之间的相互作用稳定化能E。稳定化能E越大,表示i和j的相互作用越强,即i提供电子给j的倾向越大,成键作用越强。这是通过研究占据的成键轨道和非占据的反键轨道间所有可能的相互作用,来评估它们的稳定性。计算公式为[10-11]

式中:qi——给体轨道的电子占有数;
εj、εi——对角线要素(给体和受体的轨道能量);
F(i,j)——非对角线 Fock 矩阵要素。

表3 NBO计算阿莫西林和青霉素二级微扰稳定化能Table 3 Calculated results of penicillin and amoxicillin molecule at B3LYP/6-31G(d)level by NBO analysis
表3列出了主要的稠环中的电子供体轨道i和电子受体轨道j相互作用的稳定化能。阿莫西林和青霉素相应轨道间的稳定化能基本一致,说明取代基并没有影响到环中相应轨道间的相互作用。稳定化能总体偏低,说明环上原子之间相互作用较差,环相对不够稳定。N1上的孤对电子是具有一定倾向的向C4==O13的反键轨道转移,但趋势并不明显。这进一步说明了N1与C4==O13存在较弱的共轭作用。
采用NBO的方法计算了阿莫西林和青霉素的Wiberg的键级值,见表4。通常,化学键的键级越高,相应化学键的稳定性越好。从NBO分析看,四元环和六元环中的化学键键级基本都小于标准的单键(1.0)。这说明β-内酰胺环内相应化学键不够稳定。其中,只有N(1)-C(4)的Wiberg键级值分别为1.017 4和1.017 9,处于单键(1.0)和双键(2.0)之间,但非常接近标准的单键(1.0),故可以推测N1与C4==O13的共轭极弱[11]。这说明N(1)-C(4)并没有很好的分散N上的孤对电子,N依旧容易受到亲核试剂的进攻。
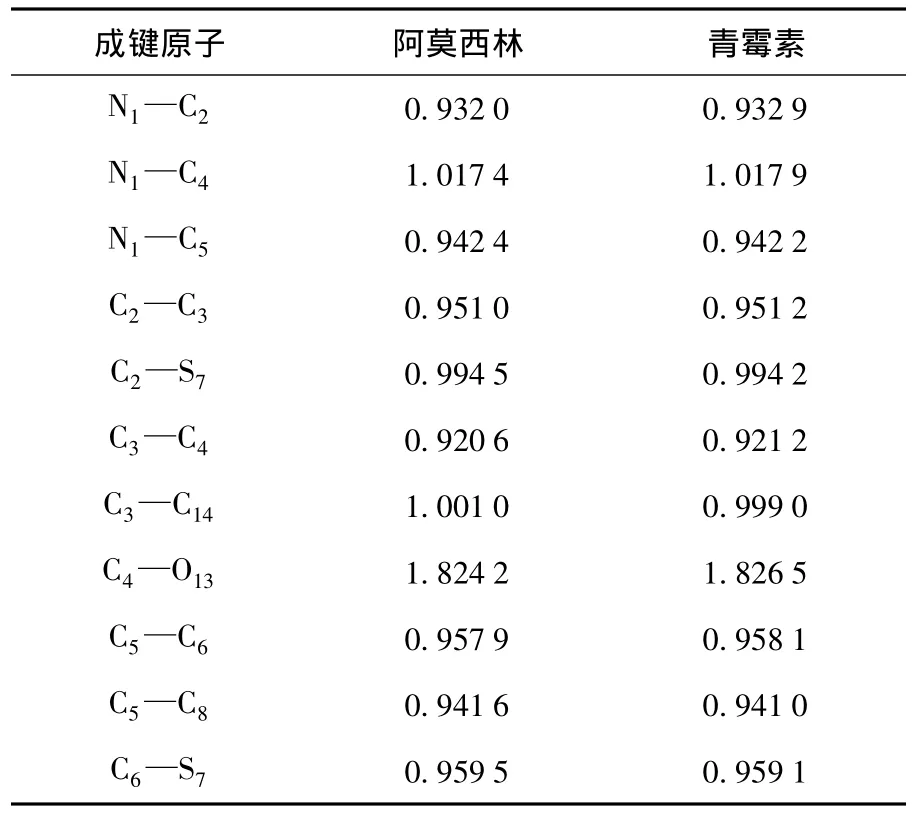
表4 6-31G(d)水平下NBO计算阿莫西林和青霉素Wiberg键级值Table 4 Wiberg bond index of of penicillin and amoxicillin at B3LYP/6-31G(d)level
2.4 前线分子轨道
阿莫西林和青霉素的HOMO和LUMO能级跃迁图如图2所示。从图2中可以看出,阿莫西林与青霉素的EHOMO均为负值,说明电子状态比较稳定。ΔE为最低未占据轨道(ELUMO)和最高占据轨道能量(EHOMO)之差,分子整体的稳定性与这些前线分子轨道能量密切相关。能极差越大,电子就越不容易跃迁,分子就越稳定。阿莫西林的前沿轨道能隙ΔE低于青霉素,可以表明青霉素比阿莫西林具有更好的热力学稳定性。两个分子的HOMO主要都位于苯环和上,是该分子中主要的电子供体部分。LUMO主要位于β-内酰胺环上,是该分子中主要的电子受体部分。
通过前期研究[12],发现极化连续介质模型B3LYP/6-31++G**的计算结果与实验检测数据具有很好的吻合性,故采用该方法计算了溶剂化效应对分子前线轨道能量的影响,并与实验结果进行了比对。通过表5发现,甲醇和氯仿对 EHOMO和ELUMO的影响较小,而水对ELUMO的影响十分明显。这主要是因为两个化合物都容易与水相互作用,形成分子间氢键。
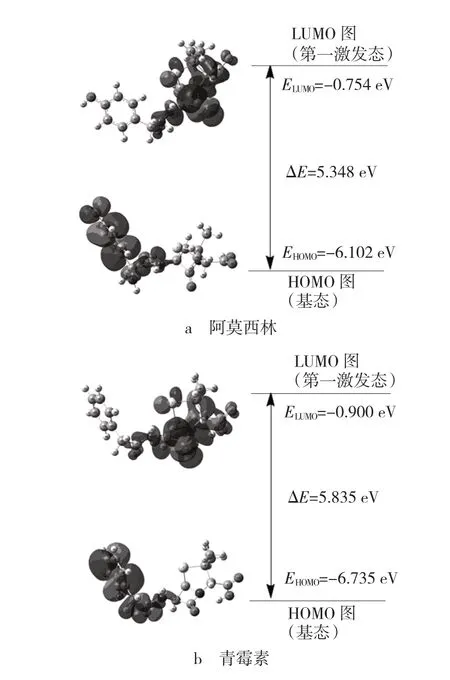
图2 6-31G(d)水平下阿莫西林和青霉素前线分子轨道Fig.2 Atomic orbital composition of frontier molecular orbital for Amoxicillin and penicillin at B3LYP/6-31G(d)level
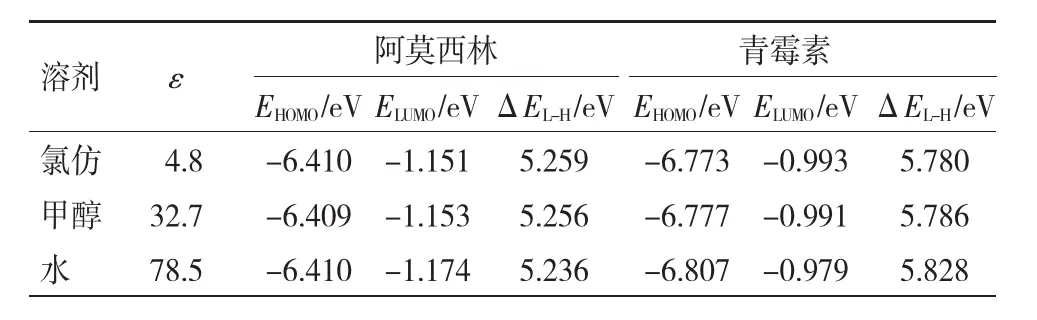
表5 6-31++G**水平下不同溶剂中青霉素、阿莫西林能带隙Table 5 Frontier orbital energies and difference for amoxicillin and penicillin in different solvents at 6-31++G** level
2.5 分子静电势
分子静电势为分子外部的单位正电荷从无穷远处移向分子与分子体系中所有粒子的相互作用势。分子所在空间的静电势为[13]

式中,V(r)是分子体系所带的电荷在r处产生的静电势,ZA是RA处原子核所带的电荷数,ρ(r')是总电子密度。分子所在右边第一项表示分子中所有原子核在r处产生的静电势,第二项是位于r'的负电荷在r处产生的静电势。
静电势图与分子的偶极矩、电负性、电荷以及化
学反应都有一定的联系,为了解分子的相对极性、形状、电荷及反应部位提供了一个非常直观的方法。阿莫西林和青霉素分子表面静电势的电子密度等值面如图3所示。其中,负电势处电子能级高、密度小(红色的区域),正电势处电子密度低(蓝色的区域)。

图3 阿莫西林和青霉素的静电势Fig.3 Electrostatic potential mapped on molecular surface
观察静电势图可以发现:两个分子中电子密度较低的区域存在着差异。阿莫西林的电子密度低的区域的空间位阻明显小于青霉素的空间位阻,阿莫西林更容易以分子间氢键的形式相互作用。取代基的引入改变了整个分子的电子排布,导致了相应化学性质的改变。
2.6 化学硬度
化学硬度是衡量系统抵抗电子分布变化能力的重要参数。其计算公式为

式中:E——体系电子能;
N——电子数;
Z——受体或供体原子数(分别用A、B表示);
μ——电子化学势;
EIP、EEA——垂直电离势和电子亲合能。
根据公式得阿莫西林和青霉素的化学硬度分别为4.283 8、4.493 6 eV。青霉素电荷变化需要更高的能量,也具有更强的抵抗电子变化的能力。硬度与轨道能量密切相关,含亲核性电子的轨道能量越低,或可接受电子的空轨道的能量越高,则硬度越大。因此,青霉素的硬度大于阿莫西林,同样青霉素的能带隙大于阿莫西林(表6)。

表6 青霉素和阿莫西林垂直电离势EIP、垂直电子亲合能EEA、化学硬度η、化学软度S、化学势μ、亲电性ω和电负性χTable 6 Predicted vertical ionization potentials,vertical electron affinities, chemical hardness, chemical potential,electronegativity,chemical softness and electronphilicity of penicillin and amoxicillin
3 结论
采用精确的杂化密度泛函理论方法,对青霉素和阿莫西林分子结构进行了几何全优化计算,得到了稳定的几何构型。在此基础上,研究其相应化学结构和性质对化合物稳定性的影响。结果表明,阿莫西林和青霉素稠环张力是导致该类化合物不稳定的主要原因。N1与C4==O13之间存在的较弱共轭作用,但N1仍然容易受到亲电试剂的进攻,容易导致β-内酰胺环开环分解。该类化合物R1和R2取代基的差异并没有影响稠环中相应的结构和性质,但取代基可以改变分子整体的电荷分布、化学硬度、能带隙。
稠环张力是导致该类化合物不稳定的主要原因,如何降低稠环的张力,提高化学稳定性,需要综合考虑药理、毒理、化学稳定性等因素,有待进一步研究。静电势图直观的表明羟基的引入明显地改变了正电荷中心部位,这对于稠环的保护以及聚合反应的控制都具有重要意义。
[1]卢昕博.青霉素G钠中致敏性杂质的分离[D].杭州:浙江大学,2009:10-13.
[2] PARK H,BROTHERS E N,MERZ K M.Hybrid QM/MM and DFT investigations of the catalytic mechanism and inhibition of the dinuclear zinc metallo-β-lactamase ccrA from bacteroides fragilis[J].J AM Chem Soc,2005,127(12):4232-4241.
[3]WOZ'NICA M,BUTKIEWICZ A,GRZYWACZ A,et al.Ring-expanded bicyclic β-lactams:a structure-chiroptical properties relationship investigation by experiment and calculations[J].J Org Chem,2011,76(9):3306-3319.
[4]BEBU A,SZABÓ L,LEOPOLD N,et al.IR,raman,SERS and DFT study of amoxicillin[J].J Mol Struct,2011,993(1/2/3):52-56.
[5]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian03,Revision A.9[CP].Pittsburgh PA:Gaussian Ine,2003.
[6]BOLES M O,CIRVEN R J,GANE P A C.The structure of amoxycillin trihydrate and a comparison with the structures of ampicillin[J].Acta Cryst,1978,B34:461-466.
[7]DEXTER D D,VAN DER VEEN J M.Conformations of penicillin G:crystal structure of procaine penicillin G monohydrate and a refinement of the structure of potassium penicillin G[J].J Chem Soc,Perkin Trans,1,1978(3),185-190.
[8]裘云锋,曹泽星.分子内结构环境对解离能的影响[J].高等学校化学学报,2008,29(12):2489-249l.
[9]张宏森,周 扬.电喷雾串联质谱法研究阿莫西林的裂解方式[J].中国药房,2010,21(29):2762-2764.
[10]周丹红,王玉清,贺 宁,等.Cu(I),Ag(I)/分子筛化学吸附脱硫的π-络合机理[J].物理化学学报,2006,22(5):542-547.
[11]牛晓庆,张建国,王 颖,等.叠氮唑类高氮含能化合物的理论研究[J].化学学报,2011,69(6):610-616.
[12]张宏森,周 扬,张 钢.精氨酸的电子结构和光谱性质[J].黑龙江科技学院学报,2010,20(3):206-210.
[13]ZHUROVA E A,MATTA C F,WU N,et al.Experimental and theoretical electron density study of estrone[J].J AM Chem Soc,2006,128(27):8849-8861.
Electronic structures and chemical stability of penicillin and amoxicillin
ZHANG Hongsen1,2, YANG Xueshun1, ZHANG Gang3, XIE Liping1
(1.Modern Analysis& Research Center,Heilongjiang Institute of Science& Technology,Harbin 150027,China;2.College of Materials Science& Chemical Engineering,Harbin Engineering University,Harbin 150001,China;3.State Key Laboratory of Theoretical and Computational Chemistry,Institute of Theoretical Chemistry,Jilin University,Changchun 130023,China)
Chemical stability of penicillin antibiotics has been challenge to theoretical studies and pharmaceutical production.This paper introduces the selection two kinds of representative drugs,namely penicillin,amoxicillin and the application of the hybrid density functional theory methods to an analysis of the effect on chemical stability by chemical structure in everything from molecular geometry,bond dissociation energy,natural bond orbital,and electrostatic potential to the hydrogen bonds between molecules,and chemical hardness.The results show that the ring tension occurs as the main cause of the instability,and substituents have great influence on the electron distribution and intermolecular interactions.The study provides theoretical references for design of the penicillin antibiotics,investigation into the structure-activity relationships and the laws of chemical reactions,and control of product quality in pharmaceutical production.
penicillin;amoxicillin;density functional theory;chemical stability
O641
A
1671-0118(2012)01-0017-06
2012-01-07
黑龙江省教育厅科学技术研究项目(12511472)
张宏森(1974-),男,黑龙江省齐齐哈尔人,高级工程师,博士研究生,研究方向:仪器分析检测和有机物理化学,E-mail:zhanghongsen666@sina.com。
(编辑 晁晓筠)
猜你喜欢
广州化工(2022年19期)2022-11-09
广州化工(2022年18期)2022-10-22
中国食品(2020年18期)2020-10-15
硅酸盐通报(2020年1期)2020-02-25
临床医药文献杂志(电子版)(2017年11期)2017-05-17
国外医药(抗生素分册)(2016年5期)2016-07-12
中国兽医杂志(2016年5期)2016-06-27
中国现代药物应用(2016年1期)2016-03-04
海南师范大学学报(自然科学版)(2015年2期)2015-12-23
中国药业(2014年24期)2014-05-26
