
我国黄淮麦区10个短体线虫样品种类的分子鉴定
2018-08-17 02:10刘海璐王暄李红梅李艳霞薛博文马居奎
中国农业科学 2018年15期
刘海璐,王暄,李红梅,李艳霞,薛博文,马居奎
我国黄淮麦区10个短体线虫样品种类的分子鉴定
刘海璐,王暄,李红梅,李艳霞,薛博文,马居奎
(南京农业大学植物保护学院/农作物生物灾害综合治理教育部重点实验室,南京 210095)
【目的】短体线虫(spp.)是植物根系内迁移性寄生线虫,可引起许多作物的根腐线虫病,给世界农业生产造成了极大的危害。为明确我国黄淮麦区与禾谷孢囊线虫复合侵染小麦的短体线虫种类,本研究对采自黄淮4省麦区的10个短体线虫样品进行种类的分子鉴定,分析种群系统进化关系及种内遗传变异,以期为我国小麦根部线虫病害的综合防治提供指导。【方法】对采自江苏、安徽、河南和山东4省小麦孢囊线虫发病田中的10个小麦短体线虫样品进行线虫分离,从各样品中随机挑取5条短体线虫,分别提取单条线虫DNA,扩增rDNA 18S片段并进行测序比对,选取序列有代表性的2个DNA样本进一步扩增其rDNA 28S D2-D3区以及mtDNA-COI基因片段,经序列比对分析后,利用MEGA4.0软件采用邻接法分别构建基于rDNA 18S、28S D2-D3和mtDNA-COI序列的系统进化树,通过聚类关系及相似度分析确定线虫种类,同时利用种特异性引物进行验证。【结果】扩增挑取的50条短体线虫的rDNA 18S区片段,测序得到片段长度在857—935 bp,BLAST比对分析揭示部分样品可能为短体线虫的混合种群;进一步测定的20个代表性DNA样本的rDNA 28S D2-D3区和mtDNA-COI基因的片段长度分别在771—784 bp和415—417 bp;系统进化树以及相似度分析揭示我国黄淮流域4省麦区10个短体线虫样品中有咖啡短体线虫(e)、落选短体线虫()和斯克里布纳短体线虫(),其中,江苏沛县样品JS2和山东潍坊样品SD1是落选短体线虫单一侵染样品,河南永城样品HN2和安徽淮北样品AH3是咖啡短体线虫单一侵染样品,安徽萧县样品AH2、AH5和淮北样品AH4以及河南永城样品HN1和HN3均为落选短体线虫和咖啡短体线虫的混合侵染样品,江苏徐州样品JS1是落选短体线虫和斯克里布纳短体线虫的混合侵染样品。用SCAR特异性引物扩增20个短体线虫单条DNA样本,结果显示,用落选短体线虫特异性引物PNEG-F1/D3B5能够从JS1-2、JS2-1、JS2-2、AH2-2、AH4-1、AH5-1、HN1-2、HN3-2、SD1-1和SD1-2等10个样本中扩增出140 bp的单一条带,用咖啡短体线虫引物PC1/PC2能够从AH2-1、AH3-1、AH3-2、AH4-2、AH5-2、HN1-1、HN2-1、HN2-2和HN3-1等9个样本中扩增出630 bp的单一条带,用斯克里布纳短体线虫引物PsF7/PsR7从JS1-1中扩增出130 bp的单一条带,种类鉴定结果与上述序列分析结果相一致。【结论】我国黄淮流域4省小麦孢囊线虫发病田中的短体线虫种类有咖啡短体线虫、落选短体线虫和斯克里布纳短体线虫,其中落选短体线虫是优势种,证实了短体线虫不同种群复合侵染小麦的现象较为普遍。基于mtDNA-COI基因构建的系统进化树可以有效区分短体线虫的近缘种,相比rDNA 18S和28S基因更适于作为短体线虫种类鉴定的分子靶标。
短体线虫;种类鉴定;rDNA;mtDNA;系统进化;SCAR
0 引言
【研究意义】短体线虫(Pratylenchusspp.)是一类分布广泛、寄主众多的迁移性植物内寄生线虫,可侵染植物根部组织,导致根系表皮破裂、块茎内部腐烂等;此外,短体线虫造成的根部伤口可为植物病原真菌和细菌侵入提供有利条件,诱发复合侵染,引起作物减产和农产品品质下降[1-2]。目前短体线虫属有效种已达101个[3-4],我国已报道的短体线虫有20多种,危害的作物种类包括小麦、玉米、大豆、花生、棉花、苎麻、马铃薯、山药、草莓、石榴、烟草等[5]。笔者实验室在调查我国黄淮麦区孢囊线虫发生分布过程中[6-7],发现短体线虫与孢囊线虫复合侵染小麦根系的现象普遍存在,这种复合侵染对于抗线虫品种的布局具有极大的影响,因此,明确河南、安徽、山东和江苏4省小麦短体线虫的种类,对于指导我国小麦孢囊线虫病和根腐线虫病一体化综合治理具有重要意义。【前人研究进展】国外已有大量关于短体线虫危害麦类作物的报道,种类主要包括六裂短体线虫()、卢斯短体线虫()、落选短体线虫()、穿刺短体线虫()、斯克里布纳短体线虫()、桑尼短体线虫()和玉米短体线虫()等,其中对小麦危害较大是落选短体线虫、穿刺短体线虫和桑尼短体线虫[8-9]。我国安徽、四川、广西、河南和西藏等地也有短体线虫危害小麦的报道,已知种类包括落选短体线虫、穿刺短体线虫、咖啡短体线虫(e)、玉米短体线虫、卢斯短体线虫、敏捷短体线虫()和草地短体线虫()等[10-15]。近年笔者实验室在黄淮麦区开展的田间调查中,发现同一田块存在多种短体线虫混合发生的现象比较普遍,传统的线虫形态学鉴定需要有足够数量的成虫用于测计和形态学特征比较,尤其当一个样品中同时存在多种短体线虫时,首先需要将不同的种区分开再依据形态特征进行鉴定,因此操作较为繁琐且鉴定周期相对较长。而分子生物学技术的应用很好的解决了上述难题,Pinochet等[16]最早利用随机扩增多态性DNA(random amplified polymorphic DNA,RAPD)技术鉴定伤残短体线虫(),Ouri等[17]利用限制性片段长度多态性(restriction fragment length polymorphism,RFLP)技术将短体线虫7个种区分开,此后,特征序列扩增区(sequence-characterized amplified regions,SCAR)[18-21]、环介导等温扩增(loop-mediated isothermal amplification,LAMP)[22]、DNA条形码(DNA barcoding)[23]等分子技术也相继应用于短体线虫的种类鉴定。其中,SCAR技术应用最为广泛,目前包括咖啡短体线虫、落选短体线虫、斯克里布纳短体线虫、卢斯短体线虫、桑尼短体线虫、玉米短体线虫、伤残短体线虫等种类均有特异性引物报道[18-21]。此外,基于核糖体DNA(rDNA)和线粒体DNA(mtDNA)保守区序列的分析,同样对短体线虫种类鉴定、系统进化及遗传变异分析具有重要意义,Subbotin等[24]比较了短体线虫的rDNA 18S和28S D2-D3区序列,证实了短体线虫的28S D2-D3区比18S具有更高的种间变异度,28S D2-D3区基因片段更适合作为分子鉴定的靶标;王金成等[25]比较分析了短体线虫rDNA的ITS区和28S D2-D3区序列,同样认为28S D2-D3区靶标更适用于短体线虫种类的检测;而Janssen等[4]通过分析rDNA 28S、ITS以及mtDNA-COI基因序列澄清了穿刺短体线虫、伪短体线虫()和铃兰短体线虫()分类地位的长期争议。当前GenBank数据库中已经积累了大量的短体线虫rDNA 18S、28S D2-D3、ITS以及mtDNA-COI 序列信息,为利用上述基因进行序列比对分析、系统发育树构建、种内遗传变异分析等提供了极大的便利。【本研究切入点】由于短体线虫近缘种间的形态特征非常相似,且种内形态变异较大[1],传统的形态学鉴定方法费时费力,对混合群体的鉴定可能会存在误差,而分子鉴定方法操作相对简单、快捷。目前国内有关小麦短体线虫种类分子鉴定的系统研究较少。本研究对采自黄淮流域河南、安徽、山东和江苏4省小麦孢囊线虫发病田的10个小麦短体线虫样品进行线虫分离,提取单条线虫DNA,分别利用rDNA和mtDNA序列比对分析、基于SCAR-PCR的种特异性引物扩增两种方法鉴定短体线虫的种类。【拟解决的关键问题】利用单条线虫DNA扩增rDNA 18S、28S D2-D3以及mtDNA-COI片段,对扩增片段进行测序和比对分析、系统进化树构建以及序列相似度分析,明确我国黄淮麦区短体线虫的种类发生情况,为今后我国小麦根部线虫病的防治提供理论指导。
1 材料与方法
1.1 样品采集与线虫分离
于2016年12月冬小麦分蘖期和2017年4—5月抽穗杨花期,对我国黄淮流域江苏、安徽、河南、山东4省小麦孢囊线虫发病田的小麦根腐线虫病发生情况进行调查,采集小麦根系及根际土壤样品共10份,具体地理信息见表1。采用浅盘法分离样品中的线虫,收集线虫悬浮液,在体视显微镜下观察线虫的形态特征。根据短体线虫的形态特征并结合文献资料核对,对样品中的短体线虫进行初步的鉴定。从各样品中随机挑取5条短体线虫,用于DNA分子鉴定。
1.2 单条线虫的DNA提取与分子片段的扩增测序
单条线虫的DNA提取参考宋志强等[26]的方法。将线虫挑入灭菌ddH2O水滴中清洗1—2次后,挑取单条线虫放入加有16 µL ddH2O和2 µL 10×PCR Buffer(Mg2+free)的200 µL PCR管中,液氮中冷冻2 min后,65℃处理1 min,重复3次;加入2 µL 10 µg·µL-1蛋白酶K,65℃温育1 h,95℃处理10 min,在-20℃条件下保存备用。
1.3 rDNA与mtDNA通用引物扩增测序
以短体线虫单条DNA样本为模板,分别采用rDNA 18S、28S D2-D3区以及mtDNA-COI等片段通用引物[27-29]进行扩增(表2),PCR反应体系:DNA模板1 µL,5 μmol·L-1的上游引物和下游引物各2 µL,2×Ex Taq Mix 12.5 µL,ddH2O补足至25 μL。扩增条件:94℃预变性4 min;94℃变性30 s,52—55℃(按引物不同)退火30 s,72℃延伸1 min,共40个循环;72℃延伸10 min。PCR产物在1.2%琼脂糖凝胶上电泳检测,扩增产物切胶回收后连接至PMD19-T载体,转化大肠杆菌DH5,经菌落PCR验证后送至南京擎科生物公司测序。

表1 黄淮麦区10个短体线虫样品的采集信息和种类鉴定

表2 本研究所用引物信息
1.4 序列比对分析和系统进化树构建
使用ContigExpress软件将获得的序列进行拼接,用BLAST进行序列比对分析后,提交GenBank获得登录序列号。从NCBI下载国内外有关短体线虫种类和群体的rDNA 18S、28S D2-D3以及mtDNA-COI序列,利用软件ClustalX1.81对所测短体线虫序列和下载的短体线虫序列进行比对分析。采用软件MEGA4.0的邻接法(neighbor-joining method)构建短体线虫的rDNA 18S、28S D2-D3和mtDNA-COI序列系统进化树,采用Boostrap值检验分支聚类的可靠性[30],进化树显示置信度>50%的数值。此外,使用MEGA4.0对鉴定的短体线虫种类与不同地理种群进行分子序列的差异度和相似度分析。
1.5 SCAR-PCR特异性引物验证
根据系统发育树及序列比对分析结果,利用种特异性引物3对[18-20](表2)对不同DNA样本进行扩增,PCR反应体系:DNA模板1 µL,5 μmol·L-1的上游引物和下游引物各2 µL,2×Ex Taq Mix 12.5 µL,ddH2O补足至25 μL。扩增条件:94℃预变性4 min;94℃变性30 s,60—66℃(按引物不同)退火30 s,72℃延伸45 s,共40个循环;72℃延伸10 min。PCR产物在1.2%琼脂糖凝胶上电泳检测,根据相应引物的扩增片段大小检测线虫的种类。
2 结果
2.1 rDNA与mtDNA通用引物扩增片段序列分析
首先用引物G18SU/R18TYL1扩增各线虫的rDNA 18S区,均得到一条约900 bp的条带(图1-A),各片段经克隆测序后获得的片段大小范围在857—935 bp,序列比对分析显示部分样本间序列差异明显,初步判断小麦短体线虫10个样品中,有6个为短体线虫混合种群侵染样品,其余可能为短体线虫单一种群侵染样品。
根据18S序列分析比对结果,从6个混合种群侵染样品中各选取序列有明显差异的线虫DNA样本2个,从4个单一种群侵染样品中各选取序列一致的线虫DNA样本2个(线虫种群代码见表1),用引物D2A/D3B扩增rDNA 28S D2-D3区,所有样本均得到一条约780 bp的条带(图1-B),测序得到的片段大小范围在771—784 bp;用引物JB3/JB5扩增mtDNA-COI区,均获得了一条约400 bp的条带(图1-C),测序得到的片段大小范围在415—417 bp。
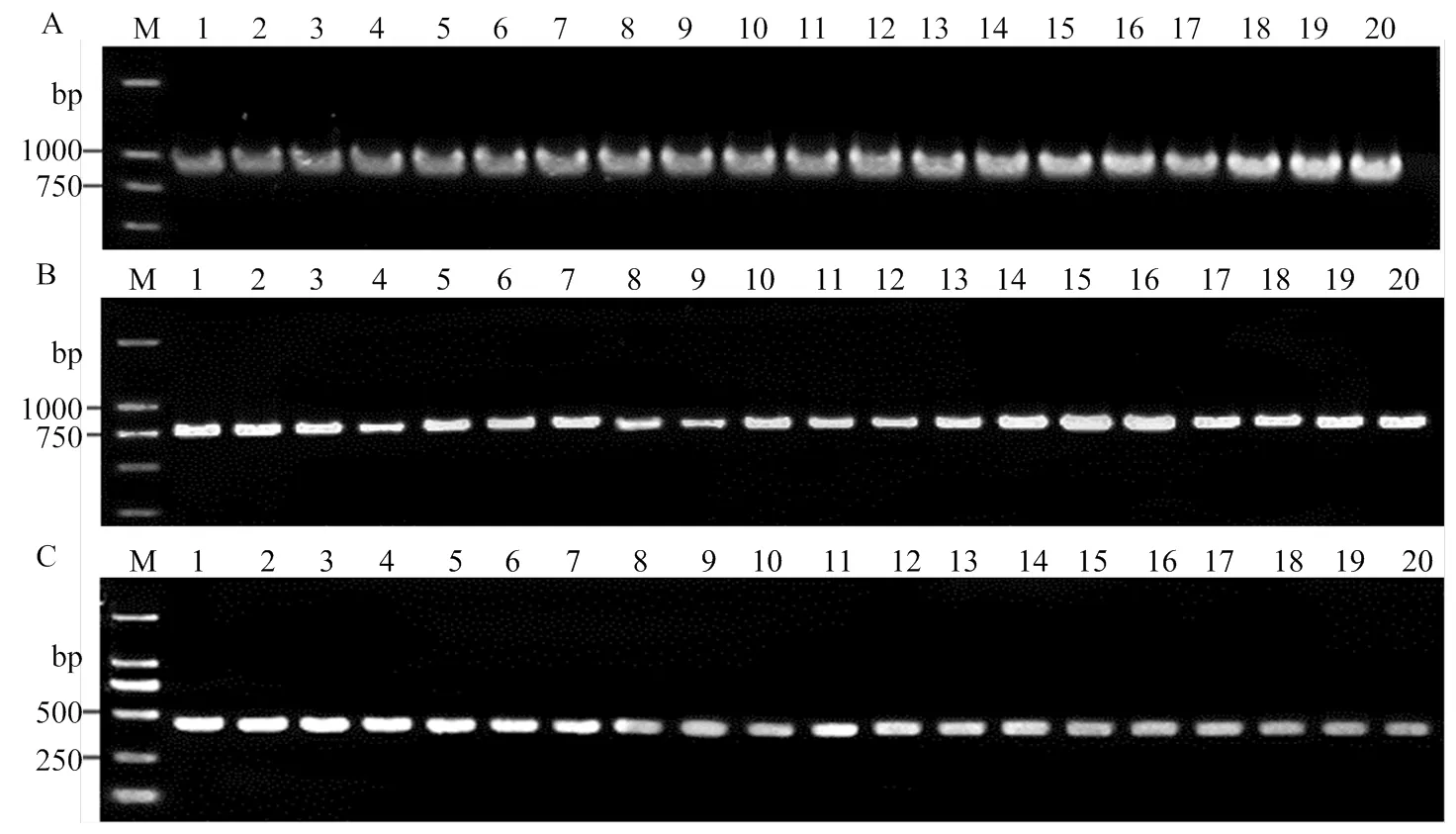
A: rDNA 18S; B: rDNA 28S D2-D3; C: mtDNA-COI.1-20: JS1-1, JS1-2, JS2-1, JS2-2, AH2-1, AH2-2, AH3-1, AH3-2, AH4-1, AH4-2, AH5-1, AH5-2, HN1-1, HN1-2, HN2-1, HN2-2, HN3-1, HN3-2, SD1-1, SD1-2; M: DNA marker DL2000
2.2 短体线虫种群的系统进化分析
从NCBI分别下载来自美国、英国、土耳其、日本、荷兰、比利时、澳大利亚、伊朗、越南和肯尼亚等国家的短体线虫不同种群的rDNA 18S、28S D2-D3和mtDNA-COI序列,与本研究获得的序列一起构建邻接法系统进化树(图2—图4),用长尾刺线虫()和细刺线虫()作为rDNA 18S树的外群,用长尾刺线虫作为rDNA28S D2-D3树的外群,用相似穿孔线虫()作为mtDNA-COI树的外群。
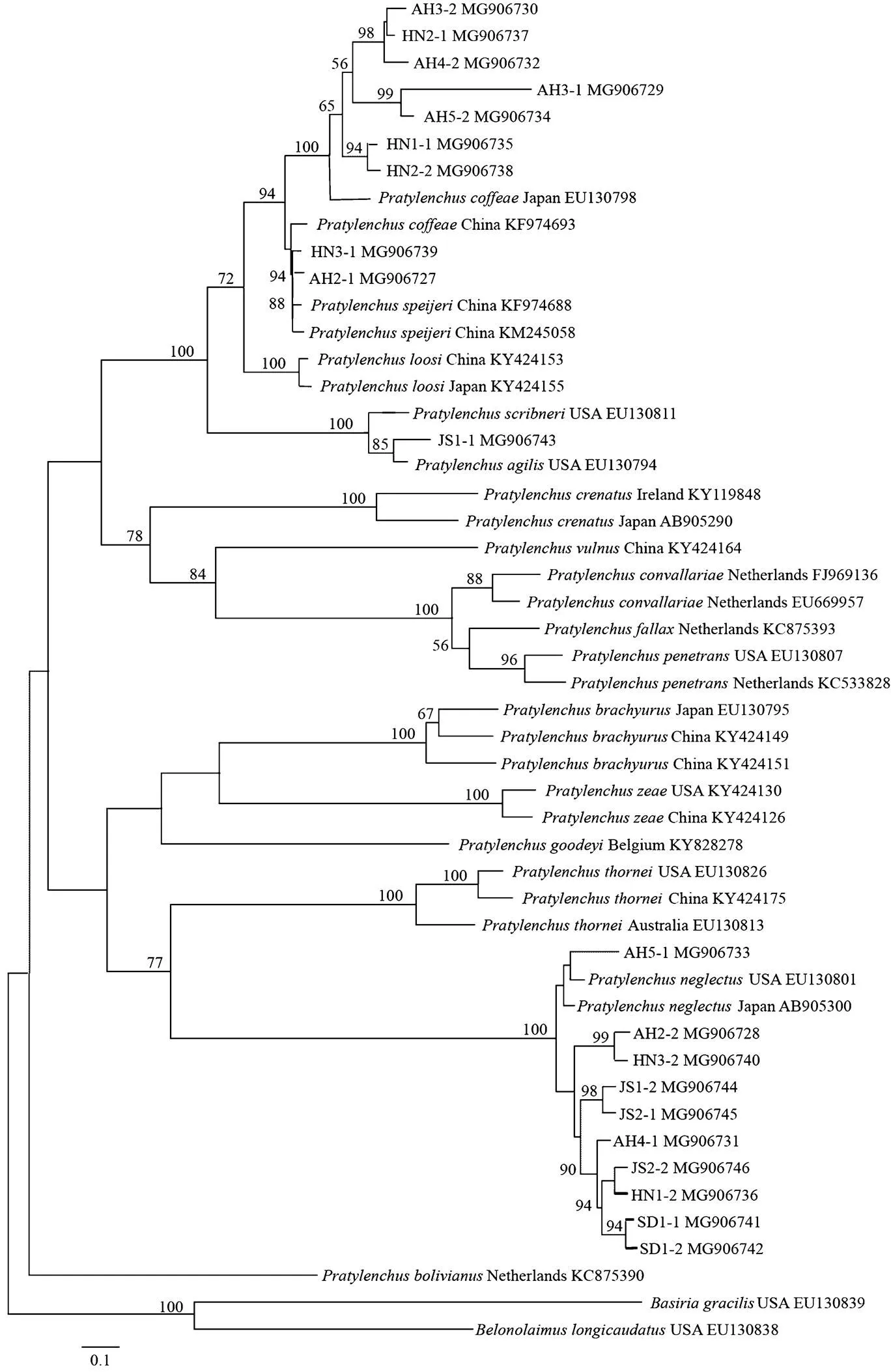
图2 基于rDNA 18S序列构建的黄淮麦区短体线虫种群邻接法系统进化树

图3 基于rDNA 28S D2-D3序列构建的黄淮麦区短体线虫种群邻接法系统进化树
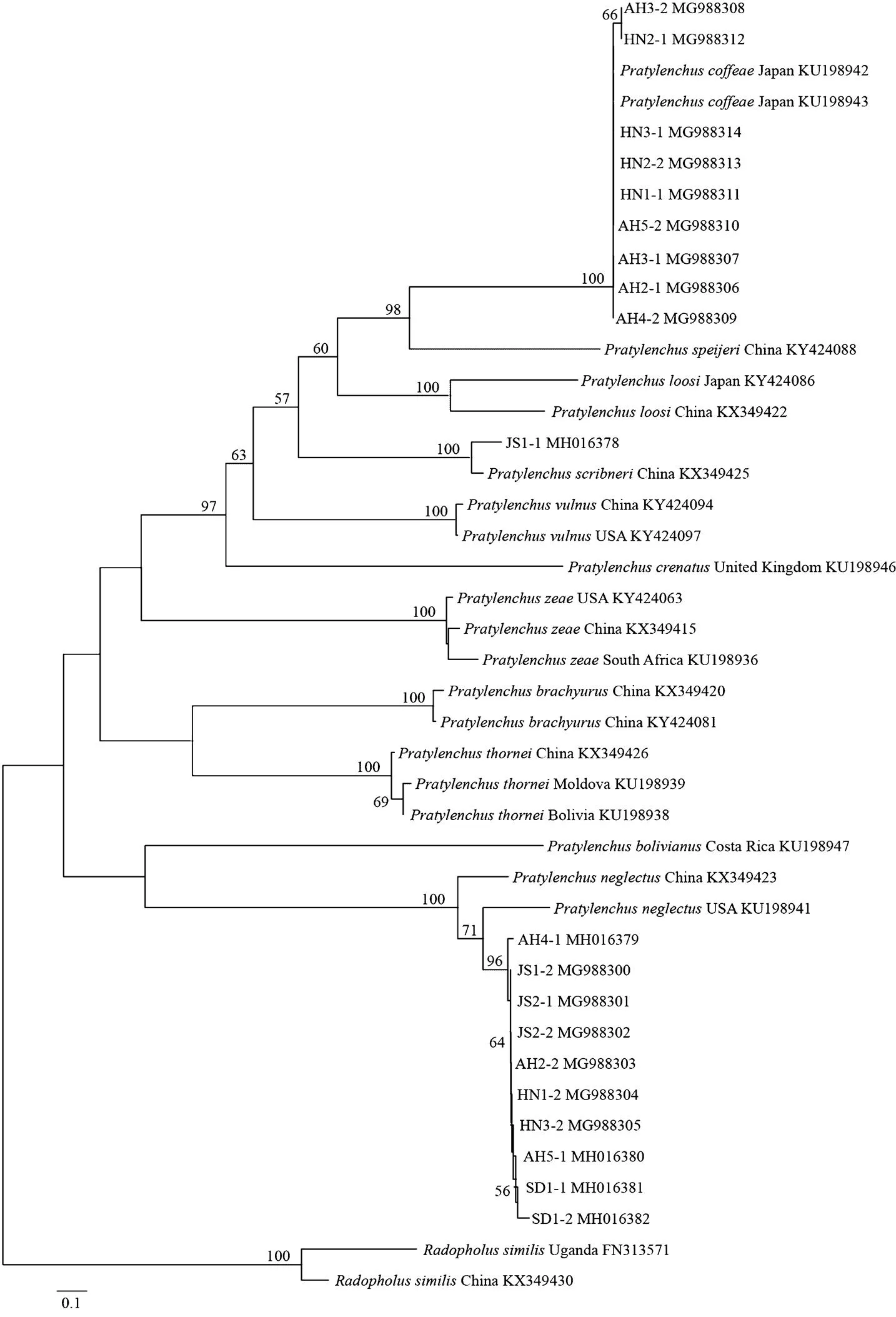
图4 基于mtDNA-COI序列构建的黄淮麦区短体线虫种群邻接法系统进化树
从rDNA 18S系统进化树(图2)可以看出,安徽小麦短体线虫种群AH2-1、AH3-1、AH3-2、AH4-2和AH5-2,以及河南小麦的短体线虫种群HN1-1、HN2-1、HN2-2和HN3-1,均与咖啡短体线虫日本、中国群体和史佩奇短体线虫()中国群体聚类在一个分支,置信度达到94%。安徽小麦短体线虫种群AH2-2、AH4-1和AH5-1,江苏小麦短体线虫种群JS1-2、JS2-1和JS2-2,河南小麦短体线虫种群HN1-2和HN3-2,以及山东小麦短体线虫种群SD1-1和SD1-2,均与落选短体线虫日本群体和美国群体聚类在一个大分支,置信度达到100%。而江苏小麦短体线虫种群JS1-1与斯克里布纳短体线虫美国群体和敏捷短体线虫聚类于一个大分支,置信度达到100%。
基于rDNA 28S D2-D3序列构建的系统进化树(图3)显示,AH2-1、AH3-1、AH3-2、AH4-2、AH5-2、HN1-1、HN2-1、HN2-2和HN3-1这9个种群均与咖啡短体线虫的美国、伊朗、越南群体聚类在一个分支,置信度达到99%,且与史佩奇短体线虫处于完全不同的进化分支;AH2-2、AH4-1、AH5-1、JS1-2、JS2-1、JS2-2、HN1-2、HN3-2、SD1-1和SD1-2这10个种群均与落选短体线虫的中国、美国、加拿大群体聚类在一个分支,置信度达到100%;而种群JS1-1与斯克里布纳短体线虫的中国、美国群体和六裂短体线虫的土耳其、比利时群体聚类在一个分支,置信度达到100%。
基于mtDNA-COI序列构建的系统进化树(图4)与rDNA 28S D2-D3进化树树形相似,20个短体线虫种群分别聚类在咖啡短体线虫、落选短体线虫和斯克里布纳短体线虫的不同分支里,置信度均为100%。
2.3 短体线虫种群的rDNA 28S和mtDNA-COI序列相似度分析
对咖啡短体线虫安徽群体AH2-1、AH3-1、AH3-2、AH4-2和AH5-2,以及河南群体HN1-1、HN2-1、HN2-2和HN3-1的28S D2-D3序列进行比对分析,结果显示,9个群体的序列差异值在0.000—0.010,相似度为99.0%—100%,它们与已知中国群体的差异值也在0.000—0.010,相似度为99.0%—100%,而与咖啡短体线虫的巴西、美国、日本、越南群体的序列差异值在0.000—0.013,相似度为98.7%—100%(表3);此外,这9个群体的mtDNA-COI序列差异值在0.000—0.003,相似度为99.7%—100%,它们与咖啡短体线虫日本群体(KU1098942和KU198943)的序列差异值同样是在0.000—0.003,相似度为99.7%— 100%(表4)。

表3 咖啡短体线虫不同群体间的rDNA 28S D2-D3 序列差异值
对落选短体线虫安徽群体AH2-2、AH4-1和AH5-1,江苏群体JS1-2、JS2-1和JS2-2,河南群体HN1-2和HN3-2,以及山东群体SD1-1和SD1-2的28S D2-D3序列进行相似度分析,从表5中可以看出,这10个群体序列间的差异值在0.000—0.020,相似度在98.0%—100%;它们与已知中国群体的差异值在0.011—0.020,相似度为98.0%—98.9%,而与落选短体线虫美国、比利时、伊朗、韩国、英国、捷克、玻利维亚群体的序列差异值同样是在0.000—0.020,相似度为98.0%—100%。此外,这10个群体的mtDNA-COI序列差异值在0.000—0.003之间,相似度在99.7%—100%,它们与落选短体线虫美国群体(KU198941)和中国群体(KY424103)的序列差异值在0.029—0.032,相似度在96.8%—97.1%(表6)。
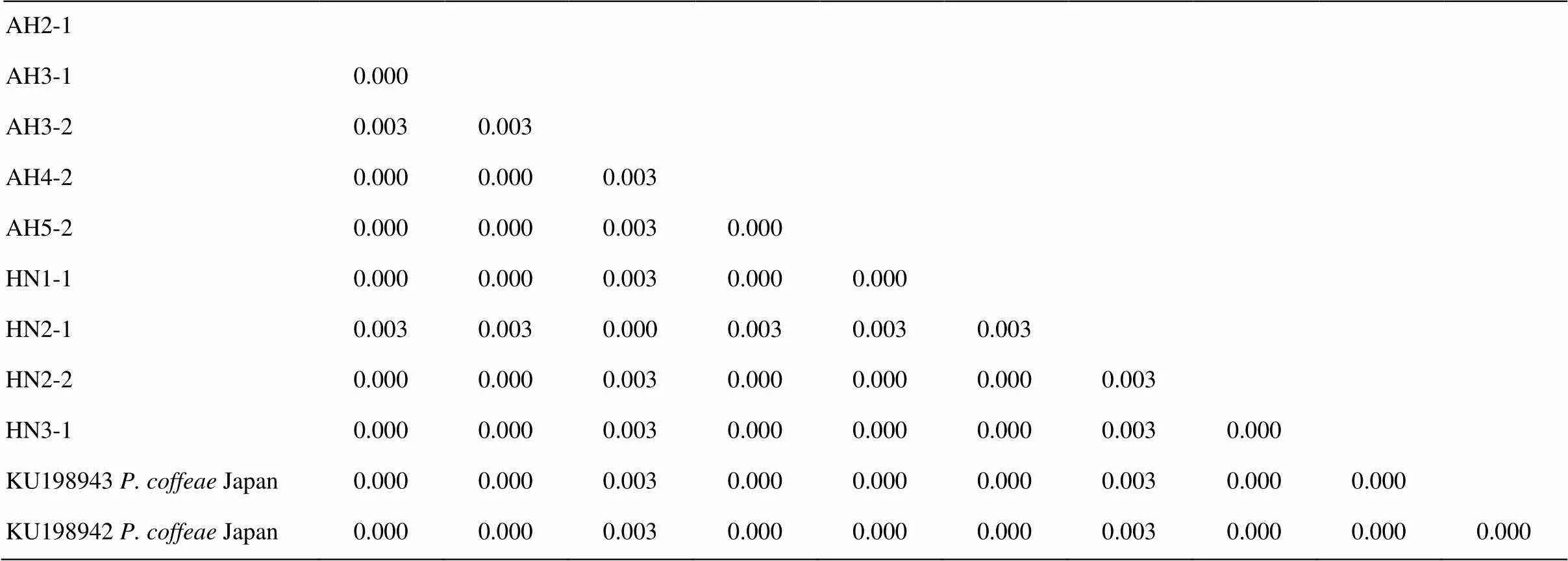
表4 咖啡短体线虫不同群体间的mtDNA-COI序列差异值

表5 不同落选短体线虫群体间的rDNA 28S D2-D3序列差异值

表6 不同落选短体线虫群体间的mtDNA-COI序列差异值
将江苏群体JS1-1的28S D2-D3序列与斯克里布纳短体线虫美国群体(KT873859、KX842628和EU130865)以及中国群体(KM094196和JX047004)的序列进行分析,结果显示它们之间的差异值在0.000—0.004,相似度为99.6%—100%;JS1-1的mtDNA-COI序列与斯克里布纳短体线虫中国群体(KX349425)的序列差异值为0.012,相似度为98.8%。
通过对黄淮流域4省10个小麦短体线虫样品的种群系统进化树及序列相似性的综合分析,揭示江苏沛县样品JS2和山东潍坊样品SD1是落选短体线虫单一种群,河南永城样品HN2和安徽淮北样品AH3是咖啡短体线虫单一种群,江苏徐州样品JS1是落选短体线虫和斯克里布纳短体线虫的混合种群,而安徽萧县样品AH2和AH5、安徽淮北样品AH4以及河南永城样品HN1和HN3,均为咖啡短体线虫和落选短体线虫的混合种群(表1)。
2.4 短体线虫种群的SCAR引物检测
利用SCAR引物对表1中的20个短体线虫种群DNA样本进行扩增,结果显示,咖啡短体线虫SCAR引物对PC1/PC2仅从AH2-1、AH3-1、AH3-2、AH4-2、AH5-2、HN1-1、HN2-1、HN2-2和HN3-1等9个样本中各扩增出一条约630 bp的条带(图5条带7、10、13所示),与预期的条带大小相一致,而落选短体线虫SCAR引物对PNEG-F1/D3B5和斯克里布纳短体线虫SCAR引物对PsF7/PsR7从上述样品中均没有扩增出条带,表明这些样本均为咖啡短体线虫。引物对PNEG-F1/D3B5仅从JS1-2、JS2-1、JS2-2、AH2-2、AH4-1、AH5-1、HN1-2、HN3-2、SD1-1和SD1-2等10个样本中各扩增出约一条约140 bp的条带(图5条带5、17、20所示),与预期的落选短体线虫扩增片段大小相一致;而引物对PsF7/ PsR7仅从JS1-1样本中扩增出130 bp的条带,与预期的斯克里布纳短体线虫扩增片段大小相一致(图5条带3所示)。SCAR检测结果表明,10个小麦短体线虫样品里20个短体线虫种群的单条DNA样本中,有9个为咖啡短体线虫,10个为落选短体线虫,仅1个为斯克里布纳短体线虫,这与上述基于rDNA和mtDNA序列分析得出的结果相一致。
3 讨论
短体线虫属不仅种类多,而且种间形态特征差异较小,种类鉴定难度比较大;此外,线虫的形态特征测计往往需要一定数量的成虫,对于群体密度较低或者混合种群的样本而言,形态测计容易产生误差,会导致依据形态特征鉴定的种类与实际情况不符。而基于线虫DNA发展起来的多种分子鉴定技术,作为形态鉴定的一种辅助手段已经广泛应用于线虫近似种的分类鉴定中[31]。尽管目前已开发了针对多种短体线虫的SCAR特异性引物[18-21],但是采用该方法需要分别使用多种引物对同一DNA样本进行扩增,如果特异性引物与样本的种类没有对应,就不会有扩增条带产生,从而无法对线虫种类进行检测鉴定。本文首先通过扩增短体线虫的rDNA 18S、28S D2-D3和mtDNA-COI片段,利用所测得的序列构建系统进化树并进行序列相似度分析,初步明确样本的种类,并在此基础上应用SCAR特异性引物进行PCR验证,成功地利用单条短体线虫DNA样本对采自黄淮流域4省的10个小麦短体线虫样品进行了种类鉴定。
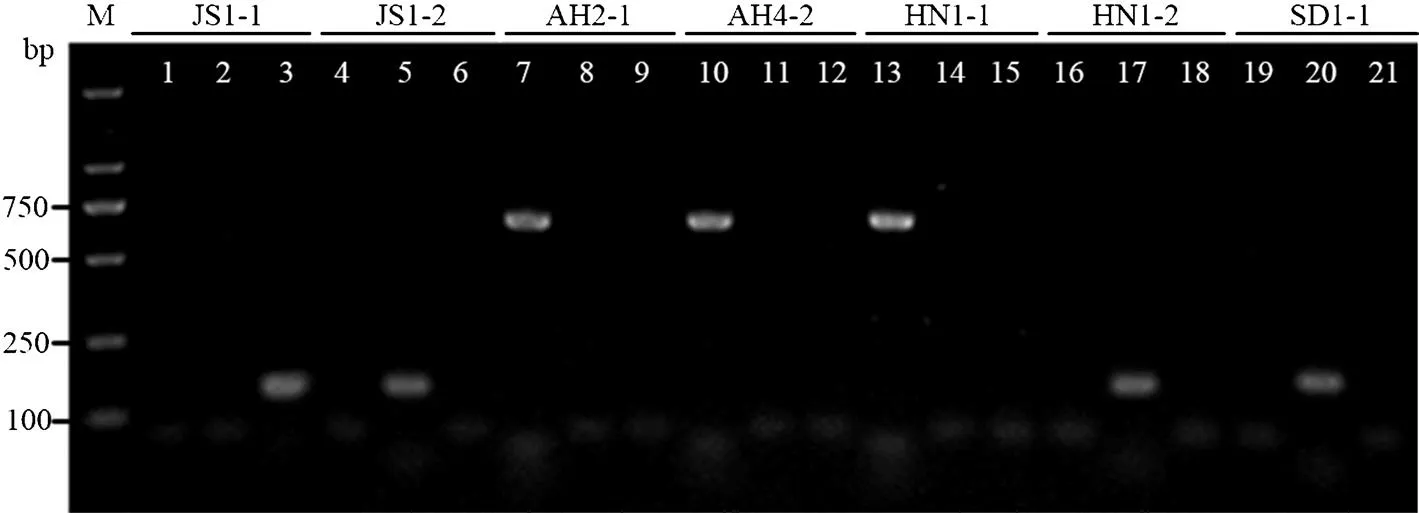
1、4、7、10、13、16、19:咖啡短体线虫SCAR引物PC1/PC2 SCAR primers PC1/PC2 for P. coffeae;2、5、8、11、14、17、20:落选短体线虫SCAR引物PNEG-F1/D3B5 SCAR primers PNEG-F1/D3B5 for P. neglectus;3、6、9、12、15、18、21:斯克里布纳短体线虫SCAR引物PsF7/ PsR7 SCAR primers PsF7/ PsR7 for P. scribneri;M:DNA marker DL2000
通过分析rDNA 18S系统进化树笔者发现,本研究测定的咖啡短体线虫群体与史佩奇短体线虫群体聚类在一个进化分支中,而在rDNA 28S和mtDNA-COI树中,咖啡短体线虫群体与史佩奇短体线虫群体聚类在不同的进化分支中。究其原因可能是由于rDNA 18S基因进化速率相对较慢,种间保守性高,更适合于属间系统进化分析[32],因此无法将咖啡短体线虫和史佩奇短体线虫区分开;rDNA 28S基因是区分动物物种及其近缘种的有效标记之一[33],在植物寄生线虫同源性比较和系统进化研究中也有广泛应用;mtDNA-COI基因由于具有单亲遗传、无重组、进化速率相对较快等特点,在分子遗传学研究中发挥着重要作用[34],也是近年来物种条形码研究的热点。通过进一步分析咖啡短体线虫与落选短体线虫rDNA 28S和mtDNA-COI序列可以看出,后者在两种短体线虫的种内遗传变异较前者更小,物种的辨识度更高。JansseN等[4]也证实mtDNA-COI分子靶标相比较rDNA 28S和ITS靶标可以更好地将穿刺短体线虫复合种群(species complex)成员穿刺短体线虫、伪短体线虫和铃兰短体线虫有效区分开,因此本文的结果也支持了mtDNA-COI分子靶标比rDNA 18S和28S分子靶标更适合于短体线虫种类的分子鉴定和系统进化研究。
李广帅[35]对河南省9个地区采集的23个小麦短体线虫样品进行了形态学和分子鉴定,认为其中有15个样品为敏捷短体线虫,其余8个样品为卢斯短体线虫。本研究对河南商丘地区采集的样品进行了鉴定,并未发现上述两种短体线虫,由于采样地点不同,无法排除不同地域线虫种类存在差异的可能性,但通过进一步的序列比对分析,笔者发现河南省郑州市惠济区敏捷短体线虫群体ZMZZ的ITS序列(JQ039330)[36]与已报道的敏捷短体线虫美国群体(FJ712889)及中国山西群体(KC952982)[37]的相似性仅为90.8%和91.0%,而中国山西群体与美国群体的相似性高达98.0%。目前的有效种地位受到质疑,被认为可能是的同种异名[38-39],本研究分析发现ZMZZ群体与斯克里布纳短体线虫江苏沛县群体JS1-1(MG906767)和美国群体(KX842626)的ITS序列相似性仅为88.9%和90.0%,由于ZMZZ群体仅报道了ITS序列,无法进一步比较分析,因此,河南ZMZZ群体的种类还有待核实与商榷。
此外,卢斯短体线虫是咖啡短体线虫复合种群(species complex)成员之一[40],形态特征与咖啡短体线虫非常相似,卢斯短体线虫区别于咖啡短体线虫的形态特征仅仅为更细的虫体,阴门位置稍偏后,尾形偏窄,尾末端窄圆或钝尖[3]。通过文献核查,笔者发现此前报道的卢斯短体线虫8个河南群体的尾形,明显宽于参考文献描绘的卢斯短体线虫尾形,且尾末端宽圆,由于这8个河南群体缺乏清晰的分子信息,因此这些河南群体的种类还有待进一步的验证。
4 结论
通过rDNA 18S、28S以及mtDNA-COI序列分析和SCAR鉴定方法,对我国黄淮流域江苏、安徽、河南、山东4省小麦孢囊线虫发病田中的10个短体线虫样品进行了种类鉴定,揭示黄淮麦区的短体线虫种类有咖啡短体线虫、落选短体线虫和斯克里布纳短体线虫,其中落选短体线虫是优势种群,短体线虫不同种群复合侵染小麦发生比较普遍。基于mtDNA-COI基因构建的系统进化树可以有效区分短体线虫的近缘种,相比rDNA 18S和28S基因更适于短体线虫种类的分子鉴定。
[1] 顾建锋, 王江岭, 何洁,陈先锋. 4种常见短体线虫形态特征比较鉴别. 植物检疫, 2013, 27(6): 69-71.
Gu J F, Wang J L, He J, Chen X F. Morphological characters of four commonspecies., 2013, 27(6): 69-71. (in Chinese)
[2] 赵立荣, 何日荣, 武目涛,胡学难, 王卫芳.3种截获短体线虫的形态与分子特征研究. 植物检疫, 2014, 28(4): 50-54.
Zhao L R, He R R, Wu M T, Hu X N, Wang W F. Morphological and molecular characterization of threenematodes imported from Japan., 2014, 28(4): 50-54. (in Chinese)
[3] Geraert E.. Gent. Belgium: Academia Press, 2013.
[4] Janssen T, Karssen G, Orlando V, Subbotin S A, Bert W. Molecular characterization and species delimiting of plant-parasitic nematodes of the genusfrom thegroup (Nematoda: Pratylenchidae)., 2017, 117: 30-48.
[5] 李广帅, 施艳, 崔娟, 杜鹃, 邢小萍, 王振跃. 小麦根腐线虫发生规律及其对小麦产量的影响. 河南农业科学, 2011, 40(4): 93-97.
Li G S, Shi Y, Cui J, Du J, XinG X P, Wang Z Y. Occurrence law of wheat root lesion nematode and its impact on wheat yield., 2011, 40(4): 93-97. (in Chinese)
[6] 王暄, 孙成刚, 方亦午, 向桂林, 刘炳良, 宋志强, 高菲菲, 李红梅. 基于GIS的小麦孢囊线虫病在江苏省的发生分布与群体密度分析. 植物病理学报, 2012, 42(5): 515-524.
Wang X, Sun C G, Fang Y W, Xiang G L, Liu B L, Song Z Q, Gao F F, Li H M. Analyses of distribution and population density of cereal cyst nematodes on wheat in Jiangsu Province based on GIS., 2012, 42(5): 515-524. (in Chinese)
[7] 刘荣荣, 王暄, 李红梅,王珍, 刘海璐. 河南省小麦主产区菲利普孢囊线虫与禾谷孢囊线虫发生情况调查.植物保护, 2017, 43(5): 157-163.
Liu R R, Wang X, Li H M, Wang Z, Liu H L. Survey of the occurrence ofandon wheat in main growing area in Henan Province., 2017, 43(5): 157-163. (in Chinese)
[8] Nicol J M, Rivoal R, Taylor S, Zaharieva M. Global importance of cyst (spp.) and lesion nematodes (spp.) on cereals: yield loss, population dynamics, use of host resistance and integration of molecular tools., 2003, 2: 1-19.
[9] 王振跃, 崔娟, 杜鹃, 李洪连, 崔长富, 崔丽娜. 禾谷类作物根腐线虫病研究进展. 河南农业科学, 2009, 38(12): 18-21, 25.
Wang Z Y, Cui J, Du J, Li H L, Cui C F, Cui L N. Advance in root lesion nematodes on cereal crops., 2009, 38(12): 18-21, 25. (in Chinese)
[10] 杨荣铮, 陈建军, 梁文慧, 陶卫平. 安徽省午季作物寄生线虫种类的调查. 安徽农业大学学报, 1983, 10(1): 91-96.
Yang R Z, Chen J J, Liang W H, Tao W P. Investigation of parasitic nematode species on summer crops in Anhui Province., 1983, 10(1): 91-96. (in Chinese)
[11] 李笃肇. 四川省植物寄生短体线虫种的记述. 西南农学院学报, 1985, 7(2): 49-51.
Li D Z. Descriptions of some species of parasitic nematodes of genuson plant roots in Sichuan Province., 1985, 7(2): 49-51. (in Chinese)
[12] 方羽生, 尹淦鏐. 植物病原线虫短体属种类的研究. 华南师范大学学报(自然科学版), 1994(4): 32-41.
Fang Y S, YiN G L. Study on pathogenetic nematodes (: Pratylenchinae) from roots of crops in Guangxi Zhuang Autonomous Region., 1994(4): 32-41. (in Chinese)
[13] 吴慧平, 杨传广, 陈良宏, 檀根甲, 王向阳. 安徽省小麦根际线虫的鉴定和分布. 安徽农业大学学报, 2010, 37(2): 189-195.
Wu H P, Yang C G, Chen L H, Tan G J, Wang X Y. Identification and distribution of rhizosphere nematodes of wheat in Anhui Province., 2010, 37(2): 189-195. (in Chinese)
[14] 王振跃, 施艳, 李广帅, 崔娟, 杜鹃, 李洪连. 河南省小麦根腐线虫病的发生危害及病原鉴定. 植物保护学报, 2012, 39(4): 297-302.
Wang Z Y, Shi Y, Li G S, Cui J, Du J, Li H L. Occurrence, damage and pathogen identification of wheat root lesion nematode disease in Henan Province., 2012, 39(4): 297-302. (in Chinese)
[15] 于焦, 金惺惺, 秦萌, 吴文佳, 徐春玲, 谢辉.西藏农作物短体线虫种类的鉴定和描述. 华中农业大学学报, 2017, 36(5): 20-24.
Yu J, Jin X X, Qin M, Wu W J, Xu C L, Xie H. Identification and description of three species of genusfrom crops in Tibet., 2017, 36(5): 20-24. (in Chinese)
[16] Pinochet P, Cenis J L, Fernández C, Doucet M, Maruli J. Reproductive fitness and random amplified polymorphic DNA variation among isolates of., 1994, 26(3): 271-277.
[17] Ouri Y, Mizukubo T. Discrimination of sevenspecies (Nematoda: Pratylenchidae) in Japan by PCR-RFLP analysis., 1999, 34(2): 205-211.
[18] Uehara T, Mizukubo T, Kushida A, Momota Y. Identification ofandusing specific primers of PCR amplification of ribosomal DNA., 1998, 44(4): 357-368.
[19] Yan G P, Smiley R W, Okubara P A, Skantar A, Easley S A, Sheedy J G, Thompson A L. Detection and discrimination ofandin DNA extracts from soil., 2008, 92(11): 1480-1487.
[20] Huang D Q, Yan G P. Specific detection of the root-lesion nematodeusing conventional and real-time PCR., 2017, 101(2): 359-365.
[21] Al-Banna L, Ploeg A T, Williamson V M, Kaloshian I. Discrimination of sixspecies using PCR and species- specific primers., 2004, 36(2): 142-146.
[22] LiuX T, Wang H H, Lin B R, Tao Y, Zhuo K, Liao J L. Loop-mediated isothermal amplification based on the mitochondrialregion to detect,2017, 148(2): 435-446.
[23] 魏亚东, 容万韬, 赵立荣, 王金成, 黄国明, 郭京泽, 孙建华. 五种短体线虫DNA条形码鉴定方法. 华北农学报, 2013, 28(6): 136-139.
Wei Y D, Rong W T, Zhao L R, Wang J C, Huang G M, Guo J Z, Sun J H. Identification of 5species utilizing DNA barcoding method., 2013, 28(6): 136-139.(in Chinese)
[24] Subbotin S A, Ragsdalea E J, Mullens T, Roberts P A, Mundo-Ocampo M, Baldwin J G. A phylogenetic framework for root lesion nematodes of the genus(Nematoda): evidence from 18S and D2-D3 expansion segments of 28S ribosomal RNA genes and morphological characters., 2008, 48(2): 491-505.
[25] 王金成, 魏亚东, 顾建锋, 张瑞丰, 黄国明, 王暄, 李红梅, 孙建华. 基于核糖体ITS区和28S rRNA D2-D3区的短体线虫系统发育研究. 动物分类学报, 2012, 37(4): 687-693.
Wang J C, Wei Y D, Gu J F, Zhang R F, Huang G M, Wang X, Li H M, Sun J H. Phylogenetic analysis of(Nematoda, Pratylenchidae) based on ribosomal internal transcribed spacers (ITS) and D2-D3 expansion segments of 28S rRNA gene., 2012, 37(4): 687-693. (in Chinese)
[26] 宋志强, 王暄, 林宇, 迟元凯, 鞠玉亮, 李红梅. 土壤中南方根结线虫的实时荧光PCR检测和定量. 植物保护学报, 2013, 40(3): 255-260.
Song Z Q, Wang X, Lin Y, Chi Y K, Ju Y L, Li H M. Detection and quantification ofin soil sample using real-time PCR., 2013, 40(3): 255-260. (in Chinese)
[27] Chizhov V N, Chumakova O A, Subbotin S A, Baldwin J G. Morphological and molecular characterization of foliar nematodes of the genus:and(Nematoda: Aphelenchoididae) from the Main Botanical Garden of the Russian Academy of Sciences, Moscow.,2006, 14(2): 179-184.
[28] Subbotin S A, Sturhan D, Chizhov V N, Vovlas N, Baldwin J G. Phylogenetic analysis of Tylenchida Thorne, 1949 as inferred from D2 and D3 expansion fragments of the 28S rRNA gene sequences.,2006, 8(3): 455-474.
[29] Derycke S, Vanaverbeke J, Rigaux A, Backeljau T, Moens T. Exploring the use of cytochrome oxidase c subunit I (COI) for DNA barcoding of free-living marine nematodes., 2010, 5(10): e13716.
[30] Saitou N, Nei M. The neighbor-joining method, a new method for reconstructing phylogenetic trees., 1987, 4(4): 406-425.
[31] 王宁, 顾建锋, 王暄, 李红梅. 进境鸡爪槭中日本短体线虫的鉴定. 南京农业大学学报, 2014, 37(4): 76-82.
Wang N, Gu J F, Wang X, Li H M. Identification ofintercepted infrom Japan., 2014, 37(4): 76-82. (in Chinese)
[32] Holterman M,van der Wurff A,van den Elsen S,van Megen H,Bongers T,Holovachov O,Bakker J,Helder J.Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades., 2006, 23(9): 1792-1800.
[33] Sonnenberg R, Nolte A W, Tautz D. An evaluation of LSU rDNA D1-D2 sequences for their use in species identification., 2007, 4: 6.
[34] Hebert P D, Ratnasingham S, de Waard J R. Barcoding animal life: cytochrome c oxidase subunit I divergences among closely related species., 2003, 270 (Suppl. 1): S96-99.
[35] 李广帅. 河南省小麦根腐线虫病原鉴定和RAPD分析[D]. 郑州: 河南农业大学, 2011.
Li G S. The identification and RAPD analysis of wheat root lesion pathogens in Henan Province[D]. Zhengzhou: Henan Agricultural University, 2011. (in Chinese)
[36] Wang Z Y, Shi Y, Li H L, Zhang M. First report of the lesion nematode,, parasitizing wheat in China., 2012, 96(5): 773.
[37] Shi H L, Zheng J W. Morphological and molecular identification ofassociated with wheat roots from Shanxi, China., 2012, 44(4): 491.
[38] Hernández M, Jordana R, Goldaracena A, Pinochet J. SEM observations of nine species of the genusFilipjev, 1936 (Nematoda: Pratylenchidae)., 2000, 3(2): 165-174.
[39] Castillo P, Vovlas N.(). Leiden, The Netherlands: Brill Academic Publishing, 2007.
[40] De Luca F, Troccoli A, Duncan L W, Subbotin S A, Waeyenberge L, Coyne D L, Brentu F C, Inserra R N.n. sp. (Nematoda: Pratylenchidae), a new root-lesion nematode pest of plantain in West Africa., 2012, 14(8): 987-1004.
(责任编辑 岳梅)
Molecular Identification ofSpecies in 10 Samples Collected from Wheat Field in Huanghuai Region of China
Liu HaiLu, Wang Xuan, Li Hongmei, Li Yanxia, Xue Bowen, Ma Jukui
(College of Plan Protection, Nanjing Agricultural University/Key Laboratory of Integrated Management of Crop Diseases and Pests, Ministry of Education, Nanjing 210095)
【Objective】Thespecies are migratory endoparasites of plant roots, causing root lesion of many crops and great damage of agricultural production all over the world. In order to clarify the species of genusco-infection withon wheat from Huanghuai region of China, 10 samples were collected from wheat field in 4 provinces of the region and thespecies were molecularly identified. The phylogenetic relationship ofspecies and genetic variation of intraspecific populations were analyzed. The results will provide valuable information for the integrated control of nematode diseases on wheat root. 【Method】Thenematodes were extracted from 10 samples collected from wheat fields infected within Jiangsu, Anhui, Henan and Shandong provinces. Fivenematodes were randomly picked up from each sample, and the DNA of individual nematode was extracted as the template for PCR amplification. The fragments of rDNA 18S region were amplified and the PCR products were sequenced. The BLAST alignment of 18S sequences revealed the differentspecies may present in these samples. The DNA templates of two representative specimens from each sample were selected for the further amplification of fragments from rDNA 28S D2-D3 region and mtDNA-COI gene. The fragments were sequenced and the BLAST alignments were performed. The phylogenetic trees were constructed on basis of rDNA 18S, 28S D2-D3 and mtDNA-COI sequences using neighbor-joining method by MEGA4.0 software, respectively. Thespecies identification was supported by the analyses of phylogenetic relationships and sequence similarities. The species-specific primers as the sequence-characterized amplified regions (SCAR) markers were used to validate the identification. 【Result】The fragments amplified from rDNA 18S region of 50 individual nematodes were sequenced and the sizes were 857-935 bp. The BLAST searching revealed that the mixedpopulations might present in some samples. The sequence sizes of the rDNA 28S D2-D3 fragments amplified from 20 selected specimens were 771-784 bp, the sizes of mtDNA-COI were 415-417 bp. The phylogenetic analyses as well as the comparisons of sequence similarities both demonstrated that threespecies including,and,were found in 10 samples collected from wheat fields in 4 provinces of Huanghuai region. Among 10 samples, the sample JS2 from Peixian of Jiangsu Province and SD1 from Weifang of Shandong Province were infected only with, HN2 from Yongcheng of Henan Province and AH3 from Huaibei of Anhui Province were infected only with. The samples AH2 and AH5 from Xiaoxian and AH4 from Huaibei of Anhui Province as well as HN1 and HN3 from Yongcheng of Henan Province were all co-infected withand, and JS1 from Xuzhou of Jiangsu Province was co-infected with. The DNA templates from 20 representative specimens were amplified using SCAR primers. The results showed that a single band of 140 bp was amplified from JS1-2, JS2-1, JS2-2, AH2-2, AH4-1, AH5-1, HN1-2, HN3-2, SD1-1 and SD1-2 using specific primer PNEG-F1/D3B5 for, a single band of 630 bp was amplified from AH2-1, AH3-1, AH3-2, AH4-2, AH5-2, HN1-1, HN2-1, HN2-2 and HN3-1 using primer PC1/PC2 for,a single band of 130 bp was amplified from JS1-1 using primer PsF7/PsR7 forThe results of SCAR detection confirmed the species identification mentioned above.【Conclusion】The molecular identification demonstrated that three species,,and, were found in wheat fields infested withfrom four provinces in Huanghuai region, andis the dominant species. The co-infection of differentspecies occurred quiet common in wheat field from the region. The phylogenetic tree based on mtDNA-COI gene can effectively distinguish the close relatedspecies, therefore, it is more suitable to identifyspecies than rDNA 18S and 28S markers.
spp.; species identification; rDNA; mtDNA; phylogeny; SCAR
2018-02-05;
2018-05-02
国家公益性行业(农业)科研专项(201503114)、国家自然科学基金(31471751)
刘海璐,E-mail:2015102038@njau.edu.cn。通信作者王暄,E-mail:xuanwang@njau.edu.cn
10.3864/j.issn.0578-1752.2018.15.006
猜你喜欢
今日农业(2022年14期)2022-09-15
天津市教科院学报(2021年5期)2021-11-10
西北农业学报(2020年12期)2020-12-14
中国森林病虫(2019年5期)2019-10-11
智富时代(2019年8期)2019-09-23
智富时代(2019年8期)2019-09-23
小天使·一年级语数英综合(2019年8期)2019-08-27
摄影之友(影像视觉)(2017年1期)2017-07-18
浙江大学学报(农业与生命科学版)(2017年1期)2017-04-17
中国医学创新(2017年7期)2017-03-31
