
基于机器学习的RNA编辑位点预测方法综述
2019-04-24 06:12冷嘉承吴凌云
生物信息学 2019年1期
冷嘉承,吴凌云*
(1.中国科学院数学与系统科学研究院 应用数学研究所,管理、决策与信息系统重点实验室,国家数学与交叉科学中心,北京 100190; 2.中国科学院大学 数学科学学院,北京100049)
RNA合成、加工、行使功能和降解是细胞生存的关键,并在许多不同的层面进行着调控[1]。RNA合成是基因表达的第一步,转录因子调控RNA聚合酶II(Pol II)与启动子结合[2],通过一套非常复杂的操作步骤将DNA转录为前体RNA[3]。前体RNA随后被加工产生成熟mRNA、功能性tRNA和rRNA[4]。RNA的加工包括(1)加帽:将7-甲基鸟苷酸(m7G)添加到5’末端[5];(2)聚腺苷酸化:在3’末端添加poly-A尾巴[6];(3)剪接:去除内含子之后拼接外显子[7];(4)RNA编辑:修改RNA分子序列并导致蛋白质多样性[4, 8]。而RNA编辑作为RNA加工中的一环,起着至关重要的作用。
RNA编辑有很多种改变RNA序列的机制,其中涉及了碱基的插入和缺失以及碱基替换,如胞苷(C)到尿苷(U)和腺苷(A)到肌苷(I)的脱氨基化,以及非模板化的核苷酸添加和插入[3]。到目前为止,人们已经在真核生物、病毒、古细菌以及原核生物中发现了RNA编辑。所以,生物学家一直对RNA编辑保持着广泛的关注和强烈的兴趣[1-3, 8]。
在哺乳动物中已经发现了两种类型的RNA编辑。一种是由催化性类多肽载脂蛋白B mRNA编辑酶(APOBEC)催化的C至U编辑,其频率相当低。另一种是A-to-I编辑,其中腺苷通过腺苷脱氨酶(ADAR)的作用脱氨基成肌苷,其频率非常高。因为人类RNA编辑中绝大多数的编辑都属于A-to-I编辑,故A-to-I编辑在生物细胞分子机制中尤为重要。研究表明,A-to-I RNA编辑同时也与脑功能、病毒感染和人类疾病有关。例如在人体中,RNA合成和加工中的错误可能引起神经系统疾病,如三核苷酸扩张性疾病强直性肌营养不良和脆性X综合征[8]。这些均显现了RNA编辑研究的重要性与意义。
深度测序技术和生物信息学的发展使得人们可以在全局筛选A-to-I RNA编辑位点。但是目前为止,通过高通量测序数据准确识别RNA编辑事件仍然是一个巨大的挑战。现在的方法通常将短读段映射到参考基因组或转录组,然后去除相同的读段,过滤低质量读段,调用差异信息并且去除已知的单核苷酸多态性(SNP)[6, 7, 10, 15-17]。但是将大量的短读段映射到参考基因组是非常耗时的,并且只有少数流水线和计算工具可公开用于处理RNA编辑[18]。最关键的是,由于成本问题,DNA测序与RNA测序一般不会一起进行,所以很难区分新的SNP位点和RNA编辑位点。实际上,大量的RNA编辑位点已被注释为dbSNP中的SNP[19]。
针对这些现象,尤其是为了区分SNP位点和RNA编辑位点,陆续产生了许多能够较为准确预测RNA编辑位点的方法,本文对一些常见方法进行了总结(见表1),将在第三节对这些方法进行详细的介绍。
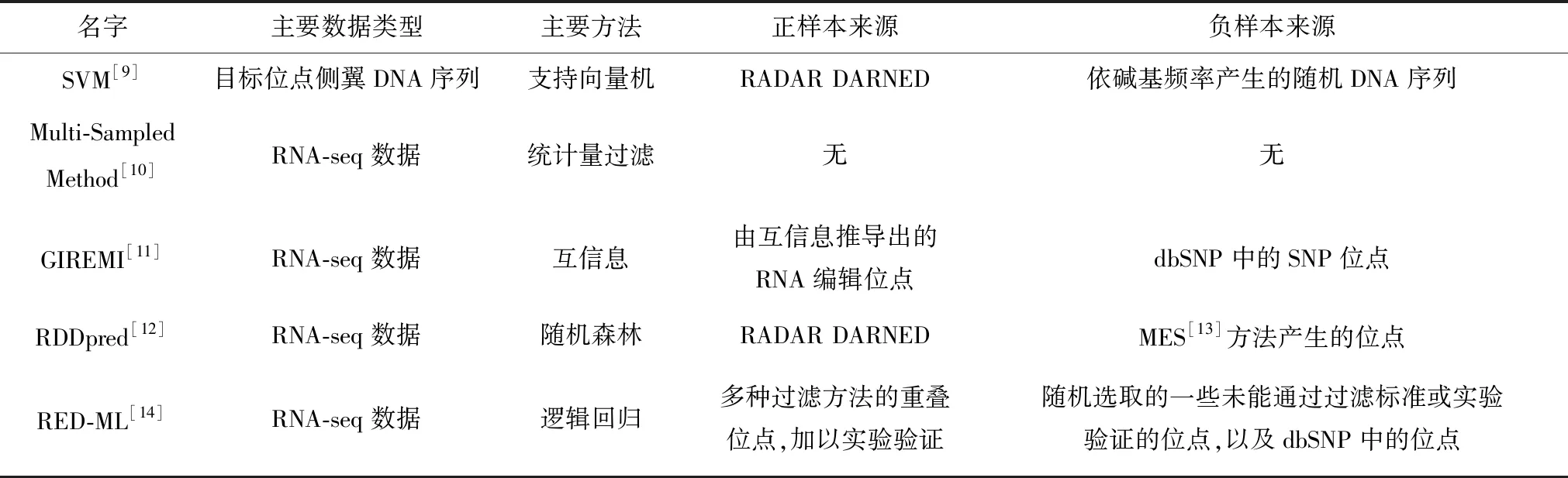
表1 RNA编辑位点预测方法概览Table 1 Overview of RNA editing site prediction methods
注:正样本指发生RNA编辑的位点(包括所有类型的RNA编辑,以A-to-I编辑为主),负样本指发生碱基变化但没发生RNA编辑的位点,其中SVM方法只针对A-to-I编辑。
1 RNA编辑数据库
已有文献在研究和评价RNA编辑位点预测方法时使用了不同的数据集,目前还没有一个通用的、被广泛接受的基准数据集。除了一些文献中提供的独立数据集,随着越来越多的RNA编辑位点被发现,目前已经出现了多个关于RNA编辑的数据库。这些数据库为研究基于计算的RNA编辑位点预测方法提供了很好的训练数据和研究的基础。
1.1 RADAR数据库
Ramaswami和Li等人于2013年创建了RADAR数据库[20],包括了人类、苍蝇、老鼠这三个物种(hg19/mm9/dm3)的A-to-I RNA编辑数据。该数据库可以根据用户的要求进行筛选,例如基因名称、是否位于Alu区域、与其他物种之间的编辑保守性等,并且提供了一些常用数据的直接下载,如hg19中的全部RNA编辑位点等。在结果中点击位置信息可以链接到UCSC基因组浏览器上浏览更详细的信息,并且都附有数据的文献来源。
1.2 DARNED数据库
DARNED数据库[21]是由Kiran和Baranov创建的。它包括多种数据来源:(1)生物信息学分析cDNA序列和基因组序列之间的差异;(2)SNP的分析数据;(3)miRNA的分析数据;(4)来自同一组织的RNA和DNA样品的高通量测序结果[21]。最后将RNA编辑事件的位点映射到参考人类基因组。此数据库不仅包括RADAR中的三个基因组还包括了hg18以及mm10。目前为止该数据库支持三种RNA编辑位点的查询方式:根据区域(染色体、位置、组织特异性等信息)查询;根据基因名称(如APOB)查询;根据序列本身查询。在此数据库的查询结果中,通过点击位置信息不仅可以链接到UCSC上,还可以链接到ENSEMBL上,比较方便。对是否与SNP混淆,与哪个SNP混淆,基因名称,基因区域都有详细的标注,而且都可以通过点击基因信息链接到NCBI上进行查询,十分人性化。
2 RNA编辑位点预测方法
2.1 基于DNA序列数据的有监督学习方法
Sun等人[9]提出了一个支持向量机(SVM)模型,基于DNA测序数据对A-to-I类型的RNA编辑位点进行预测。其核心思想是,RNA编辑位点主要是由该位点附近的序列决定的。该方法将序列与序列之间的关系进行相关性分析,得到相似矩阵,然后通过映射将其转化到内核空间得到核矩阵,最终利用SVM模型进行训练,得到具有判别能力的RNA编辑位点分类器(见图1)。
序列间的相关性是由字符串距离刻画的:
(1)
其中a,b分别代表输入的两个字符串,DEdit代表的是编辑距离,即a,b之间互相转换最少需要插入、删除或者替换多少个字符,而DHamming代表的是汉明距离,与编辑距离不同,只允许替换。而L1,L2分别代表计算编辑距离和汉明距离所用的字符串长度(见图1)。w是0到1之间取值的权重。D的值越大,代表两个序列之间的相似性越低。该方法在LIBSVM[22]中使用字符串核函数将字符串数据转换为向量空间。字符串核函数是对字符串类型数据进行操作的核函数,可写为:
K(a,b)=exp(-gamma×D(a,b)2)
(2)
其中D是从等式(1)导出的组合距离。伽玛参数定义单个训练实例影响到达的距离,低值表示“远”,高值表示“近”。
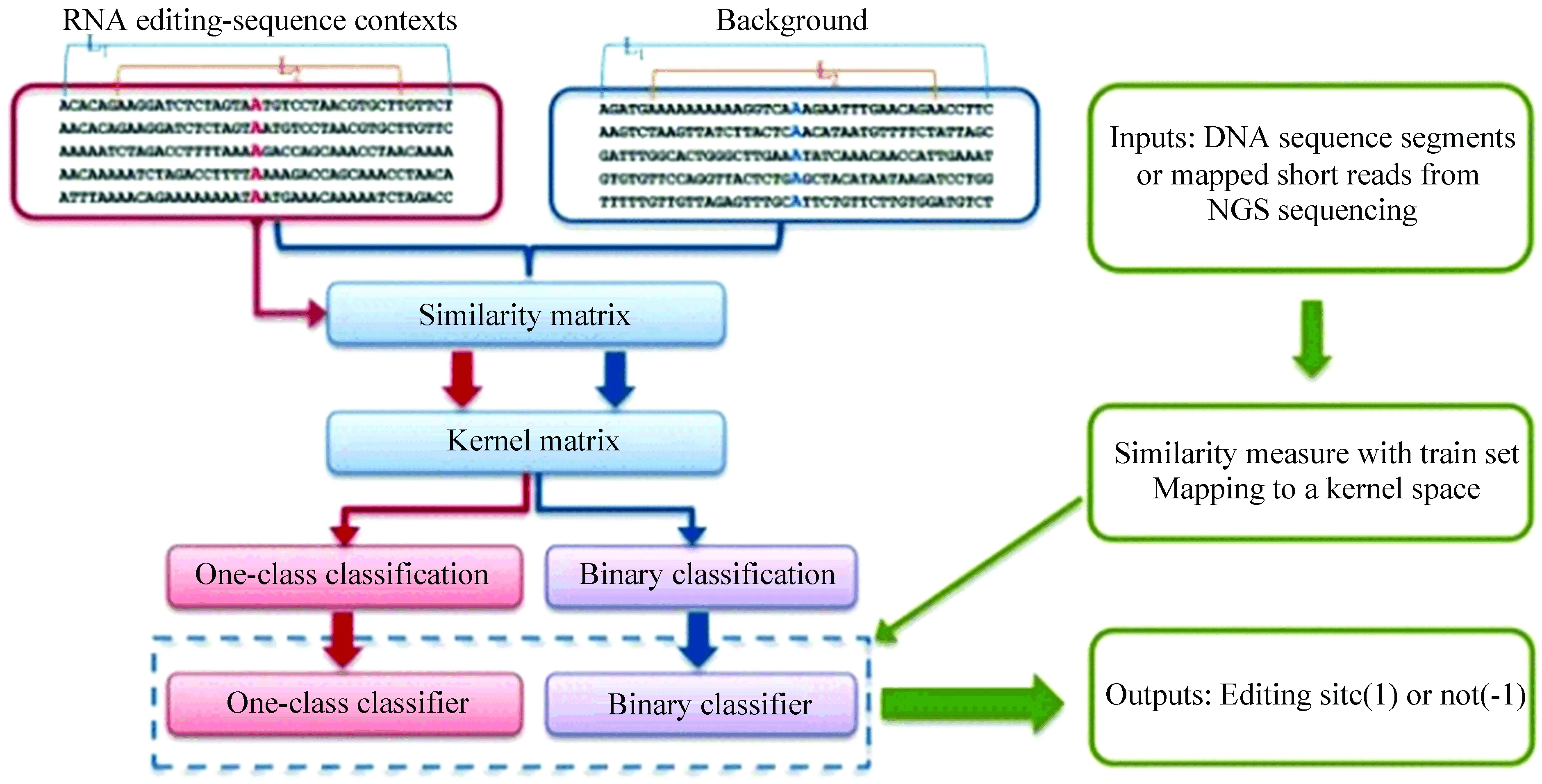
图1 SVM方法流程图[9]Fig.1 Flow chart of SVM method[9]
该论文还尝试了单类SVM。与传统的支持向量机相比,单类支持向量机尝试学习一个决策边界,实现样本与原点之间的最大分离。他们根据Schölkopf等人的研究结果[23],使用二进制值1作为编辑事件,-1作为非编辑事件。该文采用5折交叉验证的方式得到,仅用正样本作为训练数据的单类SVM的性能要远低于双类SVM,单类SVM在参数nu=0.5时的精确度在果蝇、小鼠、人类数据集上分别是0.489, 0.495, 0.498,而双类SVM模型却分别达到了0.75, 0.85, 0.79左右。并且单类SVM方法通常无法权衡在正负样本上性能的差异。该文还用基于人类样本训练的模型在一个Sanger测序集上进行了验证试验,其中79.3%(46/58)的位点被成功预测。
2.2 基于RNA-seq数据的无监督学习方法
仅仅基于DNA序列的预测方法需要可靠的RNA编辑位点数据作为训练样本,而且这类方法对于训练样本的依赖性非常高。考虑到不同物种和不同类型样本中发生RNA编辑概率的差异,以及准确的RNA编辑位点数据的缺乏,目前更为常见的是从RNA-seq数据中识别出RNA编辑位点。按照使用的机器学习方法的不同,基于RNA-seq的RNA编辑位点识别方法又可以大致分为两类:基于无监督学习的过滤方法,和基于有监督学习的机器学习方法。第一类方法通过比较RNA-seq数据和参考基因组的差异,获得了潜在的RNA编辑位点,然后通过与SNP数据库的比较以及其他指标进行过滤,去除假阳性位点,最终获得较为可靠的RNA编辑位点。而第二种方法则通过RNA-seq数据和参考基因组提取出一些特征,建立机器学习模型,利用已知的RNA编辑位点数据进行有监督训练,获得RNA编辑位点的预测模型。
Ramaswami等人[10]提出了整合多样本RNA-seq数据识别RNA编辑位点的方法。为了利用大量可公开获得的RNA-seq数据集来发现RNA编辑位点,该文章提出了两种相关且互补的方法,以使用来自单个物种中的多个个体的RNA-seq数据准确鉴定RNA编辑位点。在第一种方法(见图2(a))中,在每个RNA-seq样品中分别将测序读段映射到(非匹配的)基因组参考序列后,找出RNA的变化,并且将已知常见基因组SNP去除,以此将RNA编辑位点与其余稀有SNP区分开来。这主要是因为相同的编辑位点通常存在于不同的个体中,而罕见的SNP很可能不存在。
在第二种方法中(见图2(b)),将不同样本的RNA-seq的读段汇总到一起进行比对,从而提高找出RNA变异的灵敏度。然后如第一种方法一样,按接下来的步骤排除掉SNP,并且找出RNA编辑。由于罕见的SNP不可能出现在多个个体中,所以在汇聚后的比对中将以非常低的频率存在。

图2 Multi-Sampled Method的两种方法[10]Fig.2 Two methods of Multi-Sampled Method[10]
而GIREMI方法[11]则是典型的仅需要单组RNA-seq的方法,GIREMI将RNA-seq读段中单核苷酸变异(SNV)对的统计推断模型与机器学习方法结合,以预测RNA编辑位点。GIREMI的输入包括来自RNA-seq数据集的SNV(错配)列表和公共数据库(如dbSNP)中已知的SNP,输出是预测的RNA编辑位点及其编辑水平。除了公开的SNP信息外,GIREMI仅仅使用感兴趣的RNA-seq数据集进行所有分析,而不依赖于任何其他基因组或RNA-seq数据集,因此这种方法适用于更广的范围(见图3)。
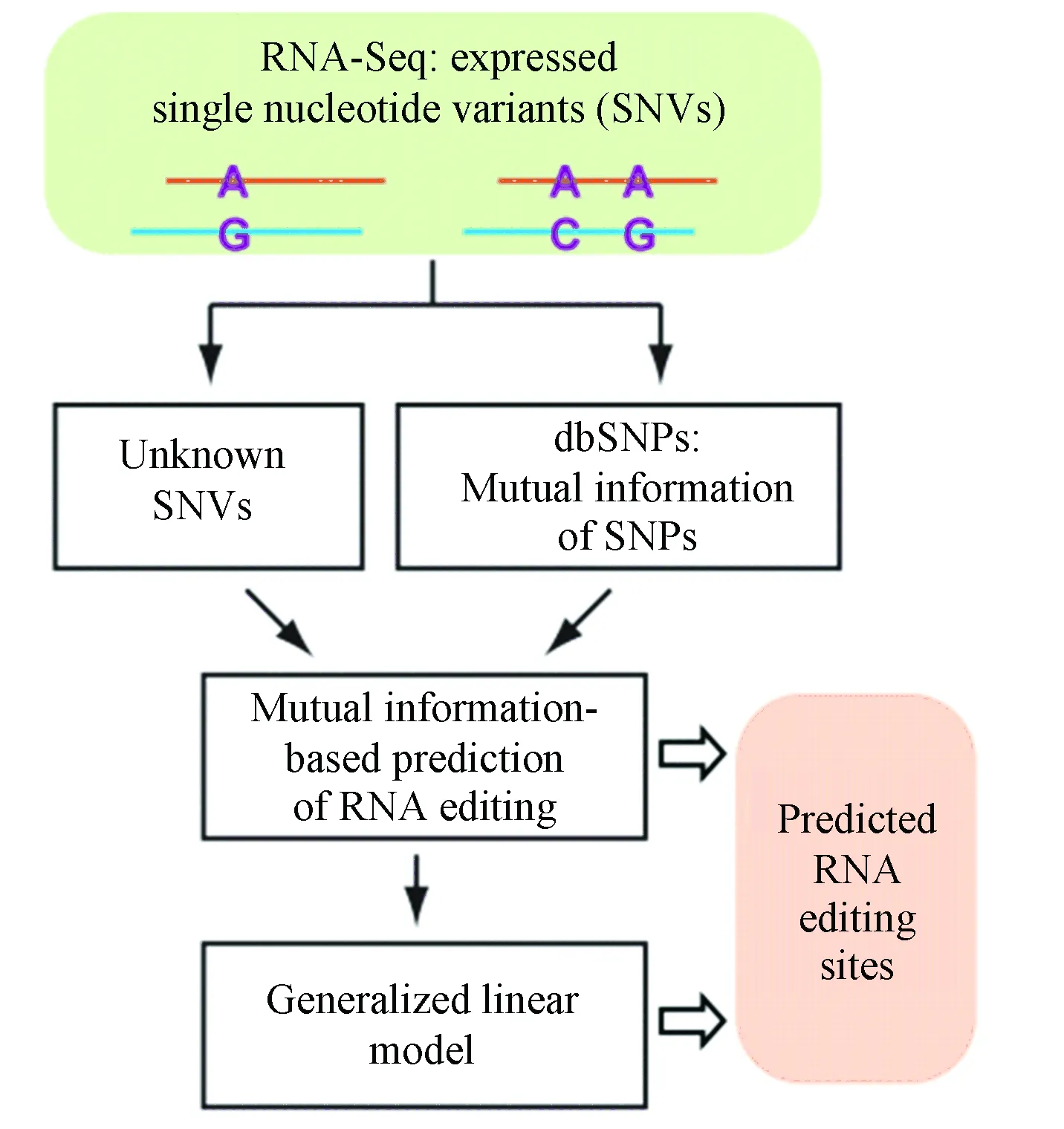
图3 GIREMI流程图[11]Fig.3 Flow chart of GIREMI[11]
GIREMI分为两个步骤,首先进行SNV位点的互信息计算:
对于每个SNV,我们考虑所有可能的碱基A,T,C,G作为变量si的四种可能状态。对于表示一对碱基(si,sj)的联合变量,总共有16种可能状态。各种状态si,sj或(si,sj)的概率可以使用最大似然法进行估计得到。考虑到所有可能的测序错误以及实际数据中的低测序深度,假设在实际数据中未观察到的状态的概率值为0.01。因此(si,sj)的互信息是:
(3)
其中N={A,T,C,G},ni和nj分别表示si和sj的状态。而SNPsi的信息值被定义为:
(4)
其中S代表带有si的SNP对的集合,T代表集合S中SNP对的个数。
这样,每个RNA-seq数据样本均产生一个基于互信息的SNP信息值分布(I(si)(见图4)。同样,对这个样本的每个SNV位点以同样的方式(SNV对)求一个信息值,取95%的置信度,如果该位点的信息值落在SNP信息分布的置信区间外,则判定该位点是RNA编辑位点,否则为SNP位点。
其次,为了提高RNA编辑位点判定的精确度,GIREMI用第一步中识别出的RNA编辑位点作为正样本来训练广义线性模型。该模型采用了两个特征,一个是读段中杂合SNP等位基因比率与SNV等位基因比率的差值的绝对值d,另一个则是基于序列本身特征的复合序列得分c(通过位置权重矩阵中+1和-1位置计算)。其回归模型为:
(5)
其中,β0,β1,β2分别为需要学习的系数,g为逻辑回归函数。
该文只对GM12878数据集进行了性能测试,并且达到了99.4%的准确度,但是其准确度的定义为100%-被预测为RNA编辑位点中SNP位点的百分比。
2.3 基于RNA-seq数据的有监督学习方法
不同于GIREMI基于自主产生的正样本来训练广义线性模型,RED-ML和RDDpred是基于已知样本进行有监督学习训练的RNA编辑位点检测模型(见图5)。

图4 SNP与RNA编辑位点的信息分布[11]Fig.4 Information distribution of SNP and RNA editing sites[11]
图5 RED-ML流程图[12]
Fig.5 Flow chart of RED-ML[12]
Xiong等人[12]建立了一个基于机器学习的RNA编辑位点预测工具RED-ML,并选择了逻辑回归(Logistic regression)进行模型的训练。RED-ML使用的特征有三大类。第一类是基本读段特征,包括候选位点的支持读段数量和计算出的编辑频率。第二类特征与可能的测序失误和错位有关,包括支持读段的图谱质量、候选位点在定位读段中的相对位置、链偏差的指示、候选位点是否落入简单重复区域等。第三类是基于RNA编辑的已知属性,如编辑类型(是否为A-to-I),候选位点是否在Alu区域以及它的序列上下文。需要注意的是,与前两类特征不同,第三类特征不能直接用来过滤非RNA编辑位点。然而,它们仍然可以提供有价值的信息,通过机器学习方法,将不同来源的信息结合起来做出分类决策。
作为一款RNA编辑检测软件工具,RED-ML的输入是一个BAM文件,也可以利用相应的基因组差异信息。RED-ML将提取候选RNA编辑位点及其相应的特征,然后应用逻辑回归分类器以相应的置信度检测真正的RNA编辑位点。RED-ML可以仅基于人类RNA-seq数据执行全基因组RNA编辑检测,也可以利用匹配的DNA-Seq数据,并与其他常见的RNA-seq数据分析步骤很好地结合。
该文用从其他文章中已发表的数据自己筛选出正负样本进行训练,其中随机选取了80%作为训练集,20%作为测试集。在ROC曲线上的AUC达到了0.98,在PR曲线上的AUC达到了0.94。并且该文在CH24T、CH62T和HeLa样本上做了RNA-seq验证实验,取阈值为0.5时成功验证了90%的RNA编辑位点。
尽管RNA-seq数据可以用于RNA编辑位点检测,但目前用RNA-seq进行RNA编辑位点检测的算法也具有相当大的假阳性(False positive)风险,这是用RNA-seq检测RNA编辑位点的最大挑战之一。由于短读段误对齐而产生的假阳性,本质上是由以下几种因素导致:(1)基因组序列固有的重复片段;(2)模糊的剪切连接;(3)个体之间普遍的多态性;(4)测序读段的短缺。RDDpred(RNA/DNA Differences prediction)[14]是一种基于随机森林算法的RNA编辑位点预测方法,能够大大减少样本中的假阳性数据,从而提升RNA编辑位点的预测准确率。
RDDpred首先对输入的测序数据进行初始比对,产生编辑位点候选者,然后从中选择满足特定条件的样本作为训练数据(见图6)。该方法使用RNA编辑数据库RADAR和DARNED中的RNA编辑位点作为正样本。而负样本则通过MES(mapping error set)方法来收集,这种方法可以在比对时计算基因组内的容易导致错误的区域[13]。从RDD候选者中收集正/负样本后,所有剩余的样本被视为预测目标。然后RDDpred建立了一个包含15个特征的随机森林预测器来预测RNA编辑位点。

图6 RDDpred 整体流程图[14]Fig.6 Overall flow chart of RDDpred[14]
RDDpred用来自Bahn和Peng的小组进行的独立研究的两个数据集进行了评估[13, 15]。在Bahn的研究中,RNA-seq产生了115 132 348个读段,RDDpred检测到6 856 440个初始RDD并预测了105 564个RNA编辑位点。在Peng的研究,RNA序列产生了583 640 030个读段,RDDpred检测到58 666 976个初始RDD并预测3 076 908个RNA编辑位点。虽然这两项研究都使用人体组织,但它们产生了不同数量的RNA编辑位点(105 564与3 076 908),这表明RNA编辑事件的表达模式在两种环境下可能不同。而同时,该方法用自己的模型验证了Bahn和Peng方法发现的编辑位点,分别成功验证了95.32%(3 947/4 141)和90.37%(20 504/22 688),并且都大幅度减少了错误的编辑位点,NPV分别达到了84.21%和75.86%。
3 讨 论
本文介绍了五种RNA编辑位点预测方法。第一种方法仅仅需要DNA序列。基于DNA序列的方法主要利用了序列间的相似性进行预测,然而其缺点就是DNA序列中包含的信息是有限的,这使得其性能没有达到较高的水准(精确度大概在0.7左右)。此外,这种方法无法用于研究RNA编辑在不同条件(例如疾病)、不同个体、不同组织中的差异。
后四类方法则基于RNA-seq测序数据和机器学习模型,它们的共同特点就是高度依赖于高通量测序的质量与深度。与此同时,还有另外一个因素也限制了这些方法的效果,那就是RNA编辑水平,如果该位点处于一个比较低的编辑水平,那么预测难度将会大大提升。
RED-ML方法提出了大量可能与RNA编辑事件有关的特征,通过逻辑回归模型进行整合。它的一个缺陷就是模型对训练数据的依赖性很高,例如用人类数据得到的模型,在其他动物上的预测效果并不理想。而且这种方法对于比对工具有很强的依赖性(目前版本只优化了BWA和TopHat2),这导致用户在选择不同的比对软件时会产生截然不同的结果阻碍研究的进行。
如何准确地区分SNP和RNA编辑,这是RNA编辑位点识别的一个核心问题。GIREMI方法通过计算不同SNV位点之间的互信息能够更准确地区分SNP和RNA编辑。但是其代价就是计算量的增加,如果考虑到时间成本的话,可能会对效率有所影响。并且要求对测序读段的长度不能太短,否则无法覆盖两个感兴趣位点。
RDDpred方法通过RNA编辑数据库和MSE对数据进行了筛选,然后用随机森林模型进行训练。该文章主要提出了组织特异性导致错误正样本的问题,因为在RADAR和DARNED数据库中有97%的编辑位点都是只存在于一个组织中,如果不清楚地将其筛选出来将会导致“预测危机”,因为这将从根本上(样本上)导致训练失败,从而降低预测性能。如果将其剔除假阳性的方法引用到其他模型中,或许会产生更好的效果,值得让人期待。
4 总 结
RNA编辑事件的识别对于理解转录后调控是非常重要的。本文首先介绍了RNA编辑的概念和意义,然后介绍了两个现有的RNA编辑数据库(DARNED、RADAR)。而随着机器学习的发展,其在RNA编辑的相关研究中也起到了重要的作用,故本文对已有的RNA编辑位点预测方法进行了概述与讨论,得到以下结论:(1)对于RNA编辑位点,我们更关注其在样本中的表达;(2)对于RNA编辑来说,仍没有一套像人类参考基因组一样较为完备的标准;(3)虽然已经有了一些基于机器学习的预测RNA编辑位点的方法,但是并没揭示RNA编辑的本质,即提取到判别RNA编辑位点的本质特征。所以RNA编辑领域的研究还有很多亟待解决的问题和现象,希望以后能够通过更深层次的模型去解释RNA编辑,从而促进相关疾病的研究以及精准医疗的发展。
猜你喜欢
上海金属(2021年6期)2021-12-02
今日农业(2021年11期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
生物学通报(2019年3期)2019-02-17
中国生殖健康(2018年4期)2018-11-06
