
小儿脓毒症休克相关基因的筛选及网络构建
2019-04-24 06:04凌家梅杨蕊萍胡迎春
生物信息学 2019年1期
凌家梅,胡 林,陈 雪,杨蕊萍,胡迎春
(1. 四川省雅安市芦山县人民医院妇产科,四川 雅安 625600;2. 四川省雅安市芦山县人民医院儿科,四川 雅安 625600;3. 西南医科大学附属医院急诊科,四川 泸州 646000;4.西南医科大学临床医学院,四川 泸州 646000)
脓毒症由是严重感染导致的全身性炎症反应[1-2],而脓毒症休克是其严重的并发症之一,其死亡率极高[3],而小儿脓毒症是儿科急诊中的危急重症,脓毒症可以引起患儿微循环障碍、免疫功能失调及组织器官功能障碍,脓毒症休克更是严重威胁着患儿的生命[4]。在脓毒症治疗方面除了早期的液体复苏、抗菌感染、以及其他的对症处理和相关并发症的防治外[5-6],还有许多新的实践,例如连续血液净化治疗的应用对于改善小儿脓毒症的急性肾损伤有一定的意义[7]。但目前对于小儿脓毒症的认知还不够深入,对疾病本身的发生、发展机制仍然不够清楚,因而没有一个有效的诊疗策略来提高该病的治愈率,或者是更为有效的方法来提前预知它的爆发,找到相应的靶点从而中断它的进程。
随着科学技术以及大数据的发展,基于生物信息学技术而进行的相关分析或许可以使脓毒症休克的发生、发展机制变得更加清晰,同时为脓毒症休克的治疗提供潜在的靶点基因,以期达到预防和治疗的目的。
通过下载GSE26378来进行标准化处理并识别出差异基因,同时进行功能富集分析及通路分析以获得小儿脓毒症休克与SIRS之间的本质区别,并建立了共表达网络和基因信号网络,通过对这些网络的构建以及核心基因的探究,旨在全局认识小儿脓毒症休克并找到一些可能的核心基因。
1 材料与方法
1.1 材料
从GEO(https://www.ncbi.nlm.nih.gov/geo/)数据库(美国国立生物技术信息中心NCBI创建的公共数据平台)下载了小儿脓毒症休克与SIRS的芯片数据GSE26378,这个芯片数据包括了29例小儿脓毒症休克与29例SIRS患儿的基因表达阵列数据。
1.2 数据处理及差异基因分析
通过将原始的数据样本发送到GCBI(https://www.gcbi.com.cn/)在线软件的分析实验室,对原始数据进行了对数标准化处理使样本芯片数据更具有可比性,并进行差异表达基因的筛选(P<0.05,FC>1.5倍)。
1.3 差异基因的GO分析
基因本体(Gene Ontology,GO)论分析是一种广泛应用于生物信息学领域的分析方法,它将筛选出来的差异基因进行简单的注释,主要包括基因的生物学功能、途径及细胞中的定位[8]。将生物学进程设置为本次分析的类型,基因显著性富集的临界参数为(P<0.05、FDR<0.05)。
1.4 信号通路分析及基因信号网络分析
KEGG是一个巨大的生物信息数据库,通过提交筛选出来的差异基因数据,应用KEGG中的功能通路分析,从而了解到差异基因的高级功能及所属的生物系统[9]。本研究找到了差异基因在KEGG功能通路中所处的位点并建立了信号通路网络,根据网络中各基因的上、下游关系筛选出可能的核心基因。
1.5 基因共表达分析
基因共表达分析是近年来比较流行的基因筛选方法,是基于基因表达特征的相似性而进行的模块分类。通过共表达分析,建立共表达网络,可以找到各差异基因表达值的相关关系,并且比较直观的发现处于核心部位的基因,为进一步的研究提供依据。
2 结果分析
2.1 差异表达基因筛选结果
通过对两组(脓毒症休克/SIRS)数据的差异表达基因筛选,共获得1 456个差异基因(P<0.05),其中下调基因859个,上调基因597个(见图1)。经火山图可知该数据上、下调基因分布基本均匀。
2.2 差异表达基因GO分析结果
差异基因进行GO功能富集分析,结果显示,top10的生物进程为:1有丝分裂细胞周期2有丝分裂细胞周期M期3细胞分裂4依赖于DNA的转录5有丝分裂中期6有丝分裂后期7蛋白质磷酸化8病毒与宿主的相互作用9细胞凋亡的过程10聚合酶Ⅱ启动子转录的正调控(见图2)
2.3 差异表达基因通路富集分析
总共有59条通路富集于网络中。主要的通路有:MAPK信号通路、P53信号通路、wnt信号通路、细胞凋亡信号通路、细胞周期受体信号通路(见图3)。
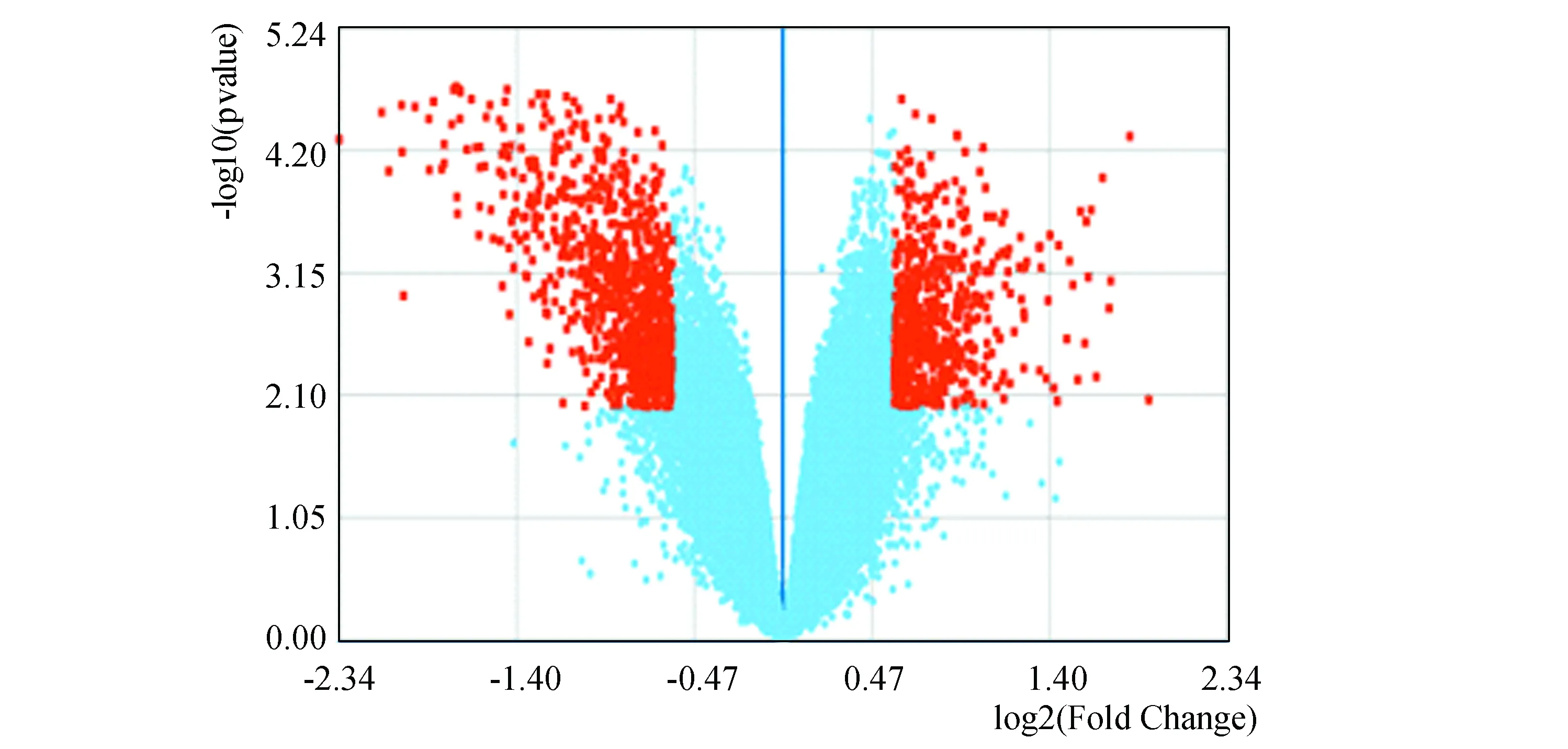
图1 差异表达基因火山图Fig.1 DEGs
注:图中横坐标为相对差异倍数(log2(FC));左侧为下调基因,右侧为上调基因;纵坐标为差异基因的显著性(-log10(P VALUE));橙色点即为筛选出的差异基因.(彩图见电子版:http://swxxx.alljournals.cn/ch/index.aspx.(2019年第1期)).

图2 GO分析Fig.2 GO analysis
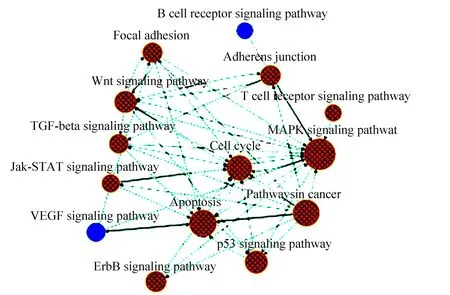
图3 通路富集网络Fig.3 Pathway analysis
注:图中圆点表示Pathway,大小代表degree值;颜色表示上下调(蓝色表示下调,黄色表示既有上调也有下调;);箭头表示上下游.
2.4 共表达分析
通过基因共表达分析,进一步揭示了差异基因表达值相关关系,并从中找到了可能的核心基因,基因CCNB1 、NUSAP1、KIF11、BUB1、BUB1B、OIP5、SHCBP1、ZWINT、FANCI、TOP2A、DLGAP5、CDKN3、NCAPG、FEN1、CCNB2等有望成为潜在的靶点。(见图4)
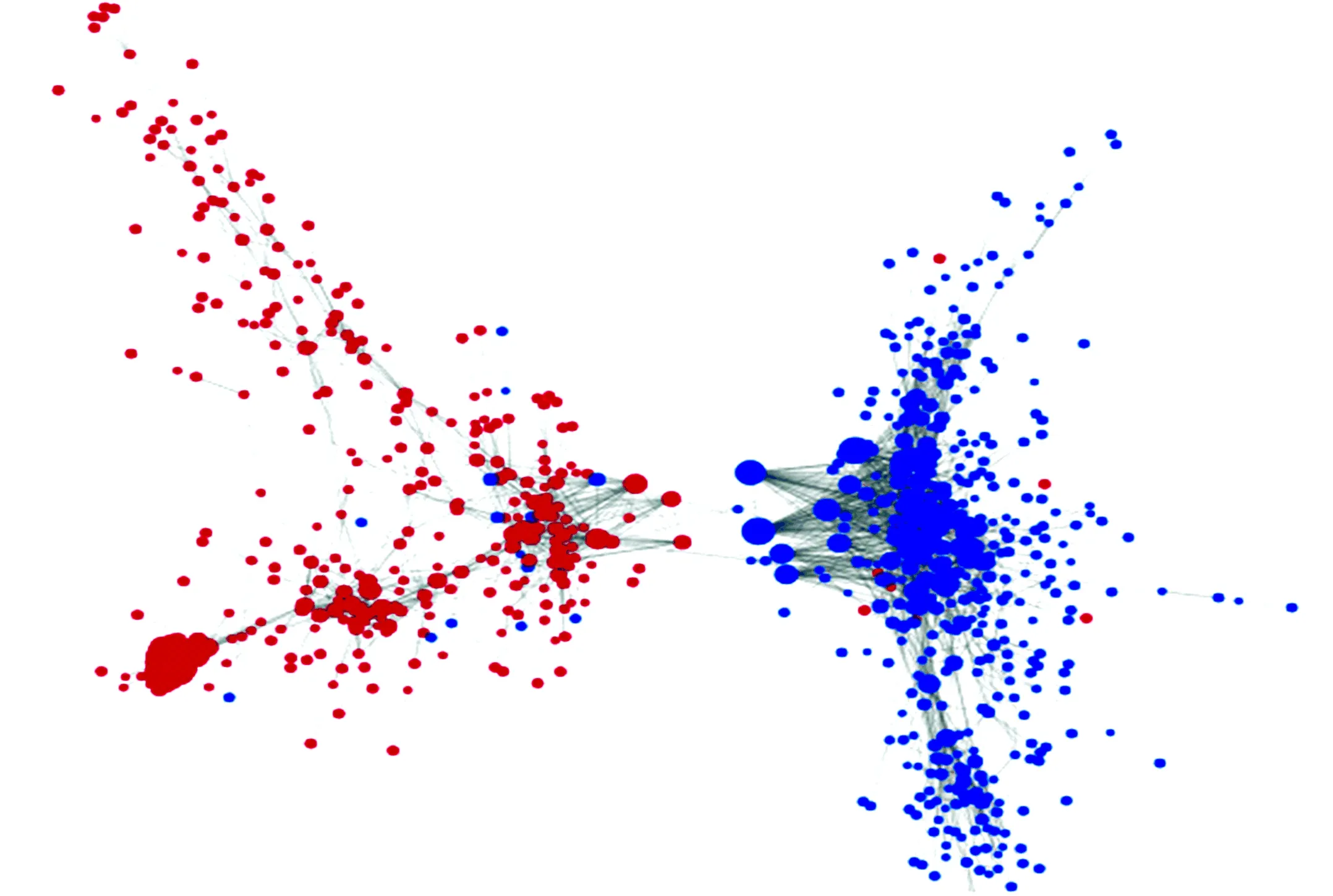
图4 共表达分析Fig.4 Co-expression analysis
注:图中圆点表示基因,大小代表degree值;颜色表示上下调(红色表示上调,蓝色表示下调);实线表示正相关,虚线表示负相关.
2.5 差异基因信号网络分析
根据KEGG数据库,基因之间具有上、下游激活、抑制等调控关系,差异基因信号网络综合分析旨在探讨差异基因的上下游调控关系,从中发现潜在的关键基因,通过网络分析发现PLCB1、PIK3R1、PIK3CA、STAT3、EP300、CAMK2D、PRKCB、CREB1、ANAPC5、CDC27等基因位于网络核心部位,有望成为新的生物标记(见图5)。
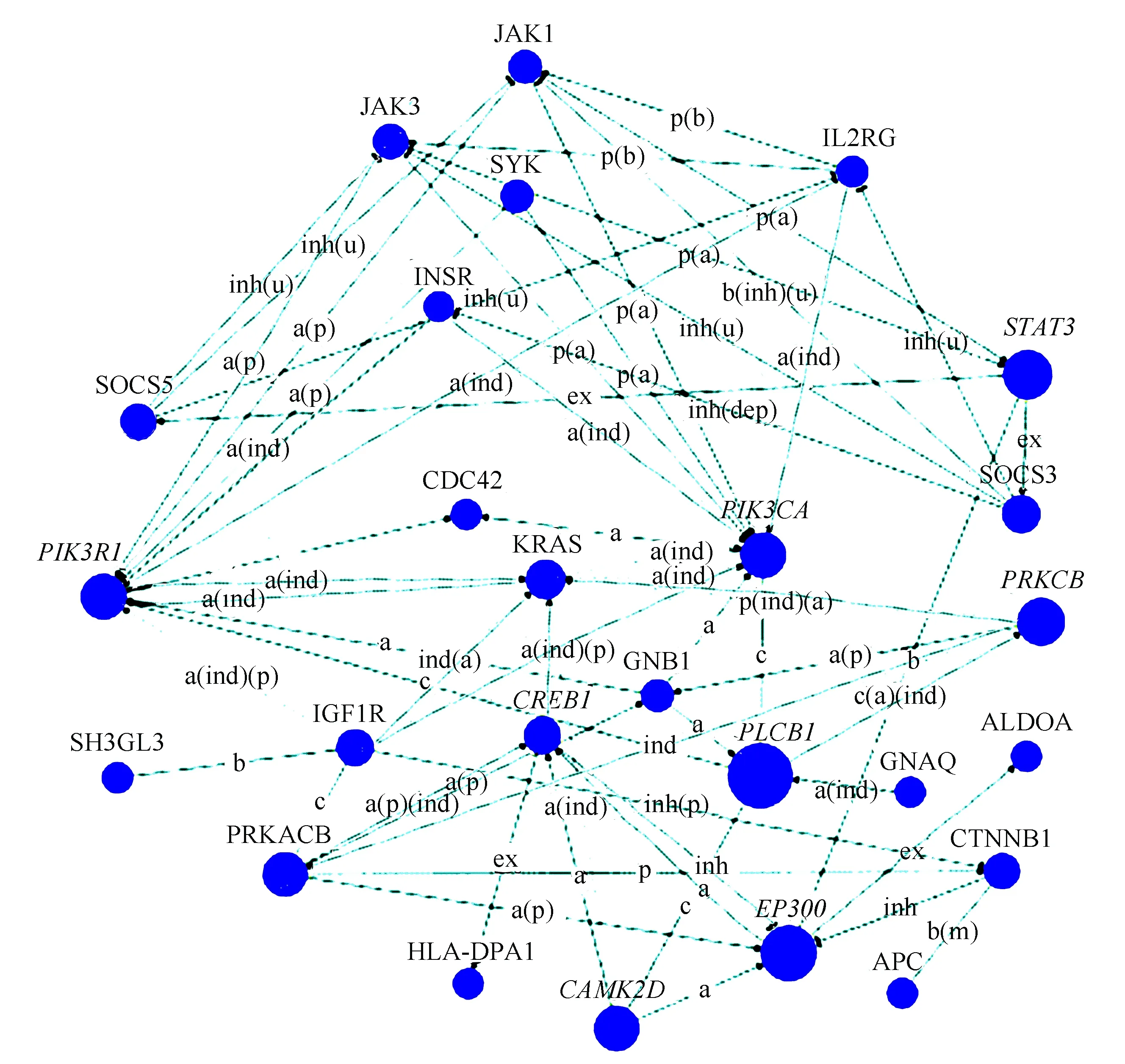
图5 基因信号网络Fig.5 Gene signal network
注:图中圆点表示基因;大小代表betweenness centrality值;颜色表示上下调(蓝色表示下调);箭头表示上下游,虚线表示间接作用.
3 讨 论
本研究通过分析了脓毒症休克与SIRS各29个样本,有1 456个基因被识别为差异基因(P<0.05),与SIRS组相比,脓毒症休克组中有条859条下调基因、597条上调基因。
将筛选出来的基因进行GO(Gene Ontology)分析,结果显示差异基因主要是富集于与细胞周期、细胞有丝分裂有关的功能模块中,这表明脓毒症休克期机体的代谢处于一个异常活跃的状态,这也与脓毒症机体炎性反应爆发、失调相符合。GO功能分析以及pathway分析都显示了,差异基因与细胞凋亡进程有关,研究证实免疫细胞的凋亡在促进了脓毒症患者免疫功能的衰竭通是也促进了器官功能障碍的进程[10]。
CCNB1、CCNB2、TOP2A共同处于基因共表达网络的核心位置,且研究证实它们参与了ARDS的发生发展的进程,是ARDS诊治过程中的核心基因,同时在本研究中,这些基因可能参与了脓毒症休克的主要进程[11]。
通过KEGG基因信号网络分析,从构建的网络模块中筛选出了处于网络核心部位的一些基因,stat3(信号转导及转录激活因子3)这种蛋白是受到细胞因子和生长因子(IFNs,、EGF、 IL5、 IL6,、HGF、 LIF 、BMP)而磷酸化激活的,它介导了细胞对于刺激时许多基因的表达,从而在细胞生长和凋亡等许多细胞过程中起关键作用。脓毒症是由感染诱发的全身炎症反应综合征,实验研究证实STAT3激活可以减弱系统性炎症反应和脓毒症的致死性,部分通过抑制免疫细胞过度活化,表明STAT3在维持宿主动态平衡中的关键作用[12]。
EP300(别名:KAT3B, RSTS2,p300)通过染色质重塑调控转录的组蛋白乙酰转移酶,在细胞增殖和分化过程中起重要作用,并通过与磷酸化CREB蛋白结合而介导cAMP基因的调节。先前的研究也已经证实P300与脓毒症中骨骼肌代谢,肌细胞流失有一定的相关性[13]。而基因NUSAP1、KIF11、BUB1、BUB1B、、PLCB1、PIK3R1、PIK3CA、CAMK2D、PRKCB、CREB1、ANAPC5是通过生物信息学技术筛选出来的相关的潜在基因,或许可以提供新的研究方向。
4 结 论
通过基因芯片技术,找到疾病相关基因,并使用生物科学技术和高通道筛选技术找到差异基因,可以更好的了解小儿脓毒症休克与SIRS之间的差异,从中筛选出可能的潜在基因,为进一步的预防和治疗策略的探索提供可能的研究方向,但需要进一步的相关实验验证。
猜你喜欢
安徽农业科学(2022年1期)2022-02-14
昆明医科大学学报(2020年12期)2021-01-26
中华养生保健(2020年4期)2020-11-16
心电与循环(2020年1期)2020-02-27
中国中医急症(2019年10期)2019-05-21
江苏农业科学(2017年5期)2017-04-15
中华老年多器官疾病杂志(2016年9期)2016-04-28
中国学术期刊文摘(2016年1期)2016-02-13
中国学术期刊文摘(2016年1期)2016-02-13
湖北农业科学(2014年3期)2014-07-21
