
注射用美洛西林钠有关物质测定方法的改进
2021-05-18 07:22牛冲窦艳丽娄红祥徐玉文
药学研究 2021年4期
牛冲,窦艳丽,娄红祥,徐玉文
(1.山东省食品药品检验研究院,国家药品监督管理局仿制药研究与评价重点实验室,山东省仿制药一致性评价工程技术研究中心,山东 济南 250101;2.山东大学,山东 济南 250014)
美洛西林钠(Mezlocillin Sodium)属于第三代半合成青霉素类抗菌药物,由德国拜耳公司研发。美洛西林主要对革兰阳性菌、革兰阴性球菌及放线菌等高度敏感,且对耐药金黄色葡萄球菌有效。主要通过干扰细菌细胞壁黏肽的合成,使之不能交联而造成细胞壁的缺损,从而导致细菌死亡。用于大肠埃希菌、肠杆菌属、变形杆菌等革兰阴性杆菌中敏感菌株所致的呼吸系统、泌尿系统、消化系统、妇科和生殖器官等感染[1-7]。
美洛西林钠收载于《美国药典》第40版(USP40)、《中国药典》2015年版(ChP.2015)(二部)[8]。USP35中未对有关物质进行控制。ChP.2015中对有关物质采用等度洗脱方法进行控制,2014年国家评价性抽验法定检验中发现,在药典规定的时间之外仍有杂质检出。彭力等[9]采用梯度洗脱方法对美洛西林钠杂质进行了研究,杂质检出的数量及杂质总量均明显增加,但其分析检验时间较长,不适合质量控制,且改进的方法中最初的等度洗脱阶段与药典方法一致,此阶段,美洛西林钠的合成中间体氨苄西林的色谱峰与溶剂峰重合,溶剂存在干扰。基于以上原因,我们通过对原料合成中间体及降解产物进行系统适用性考察,优化了测定方法,改进后的方法杂质检出能力明显提高,更好的控制产品的质量。
1 仪器与试药
1.1 仪器 Agilent 1200高效液相色谱仪(DAD检测器);岛津LC-20at高效液相色谱仪;BP211D电子天平。
1.2 试药 注射用美洛西林钠(样品来自2014年国家评价性抽验,共204批次,美洛西林钠原料、氨苄西林、氯甲酰物由山东瑞阳制药股份有限公司提供)。水为去离子水,乙腈为色谱纯。
2 方法考察
2.1 药典方法检测 色谱柱:Thermo BDS C18色谱柱(4.6 mm×250 mm,5 μm),检测波长:210 nm,流速:1.2 mL· min-1,进样量:20 μL,柱温:35 ℃;流动相A[磷酸盐缓冲液(磷酸二氢钾4.9 g和磷酸氢二钾0.45 g,加水溶解并稀释至1 000 mL)]-流动相B(乙腈)=80∶20。记录色谱图至主峰保留时间的1.5倍,色谱图见图1。可见在主峰保留时间4倍以外,仍有色谱峰检出,该方法易造成杂质的漏检,需对方法进行改进。

图1 按药典方法测定供试品溶液色谱图
2.2 色谱条件与系统适用性 洗脱方式由等度洗脱变为梯度洗脱,其余色谱条件同“2.1”项下,0~18 min,A:90%~65%;18~27 min,A:65%;27.01 min,A:90%。
供试品溶液:取本品60 mg,精密称定,加水溶解并稀释至100 mL,摇匀。
对照溶液:精密量取供试品溶液1.0 mL,加水稀释至100 mL,摇匀。
系统适用性溶液:取供试品溶液,水浴加热8 min。按上述方法测定,理论板数按美洛西林峰计为51 388,美洛西林峰与前后杂质分离度分别为2.05和1.89(见图2),系统适用性良好。
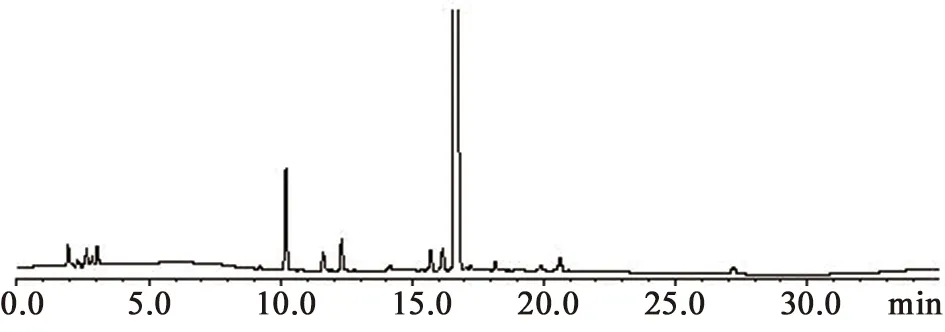
图2 系统适用性色谱图
2.3 波长的选择 采用DAD检测器对美洛西林及破坏性试验样品进行测定,美洛西林及降解产物均为紫外末端吸收,故参照ChP.2015方法,检测波长选择为210 nm。
2.4 专属性试验 溶剂无干扰。
酸破坏考察:取美洛西林钠原料(批号:14011641)60 mg,置100 mL量瓶中,加水50 mL溶解,加0.1 mol·L-1盐酸溶液5 mL,放置30 min,加入0.1 mol·L-1氢氧化钠溶液5 mL,用水稀释至刻度,摇匀。
碱破坏考察:取美洛西林钠原料(批号:14011641)60 mg,置100 mL量瓶中,加水50 mL溶解,加0.1 mol·L-1氢氧化钠溶液5 mL,放置30 min,加入0.1 mol·L-1盐酸溶液5 mL,用水稀释至刻度,摇匀。
氧化破坏考察:取美洛西林钠原料(批号:14011641)60 mg,置100 mL量瓶中,加水50 mL溶解,加30% H2O25 mL,放置30 min,用水稀释至刻度,摇匀。
光照破坏考察:取美洛西林钠原料(批号:14011641)60 mg,置100 mL量瓶中,加水溶解并稀释至刻度,摇匀,置254 nm紫外灯下放置24 h。
加热破坏:取美洛西林钠原料(批号:14011641),80 ℃加热2 h,称取60 mg,置100 mL量瓶中,加水溶解并稀释至刻度,摇匀。
照“2.2”项下色谱条件分别进样考察,色谱图见图3。上述破坏条件下产生的各降解杂质与美洛西林的色谱峰的分离度均符合要求。
2.5 定量限与检出限 精密量对照溶液(批号:14011641)3 mL,用水稀释至200 mL,摇匀,精密量取5 mL,用水稀释至100 mL,进样20 μL,以信噪比为10计算,定量限为9 ng,进样7 μL,以信噪比为3计算,检出限为3 ng。
2.6 精密度试验 取美洛西林钠(批号:14011641),于同一天内连续测定5份有关物质,单个杂质及总杂质含量的RSD分别为0.54%和0.62%,日内精密度良好。另取美洛西林钠(批号:14011641)连续5 d每天测定一份有关物质,单个杂质及总杂质含量的RSD分别为0.32%和0.24%,日间精密度良好。
2.7 稳定性考察 取美洛西林钠(批号:14011641)按“2.2”方法,分别与0、1、2、4、6 h进样测定,保留时间为10.2的杂质由0.18%增加至0.32%;从2 h开始,保留时间2.6 min处出现一杂质峰,其含量0.23%;杂质总和由2.96%增加至3.32%。提示本品不稳定,应临用现制。

A.原料(API);B.酸破坏样品;C.碱破坏样品;D.氧化破坏样品;E.加热破坏样品;F.光破坏样品图3 HPLC色谱图
2.8 1%自身对照溶液线性考察 精密量取美洛西林钠(批号:14011641)供试品溶液0.5、1.0、2.0、3.0、4.0 mL,分别用水稀释至200 mL,摇匀,按“2.2”项下方法分别进样测定,以美洛西林的峰面积Y为纵坐标,以进样量(ng)为横坐标,进行线性回归,Y=2 470.8X+7 166.7 (r=0.999 9)。结果表明,美洛西林在30.07~240.56 ng范围内呈良好线性关系。
2.9 耐用性考察 取“2.2”项下系统适用性溶液,分别采用Waters Symmery C18(5 μm, 4.6 mm×150 mm)、Welchrom C18(5 μm,4.6 mm×250 mm)色谱柱,按“2.2”项下色谱条件进样测定,美洛西林与相邻杂质的分离度均符合要求,系统的耐用性良好。
2.10 供试品测定 取供试品,照“2.2”项下方法测定。204批次样品,按药典标准检验,单个杂质在0.19%~1.27%之间,均值为0.49%,总杂质在0.44%~2.53%之间,均值为1.01%,杂质个数在6~17之间,平均10个;按改进后的方法检验,单个杂质在0.18%~1.45%之间,平均0.60%,杂质总和在0.79%~3.35%之间,平均1.98%,杂质个数在17~43之间,平均为30个。与药典结果相比,改进方法的杂质总和及杂质个数页明显高于药典方法,部分企业单个杂质变化较大,主要是因为改进方法中保留时间约为27 min的杂质峰的影响,该杂质在部分企业的产品中为最大杂质,而在药典方法中,这一杂质峰无法检出。部分供试品结果见表1。

表1 样品检测结果
3 讨论
ChP.2015方法主峰相对保留时间4倍以外仍有杂质峰检出,且随着时间的延长,峰形变宽,易造成杂质的漏检。采用改进的方法在保留时间27 min左右有一明显的杂质峰检出,此杂质在部分企业的样品中为最大杂质,从而导致样品最大杂质有较大的增加。专属性研究表明,该杂质在各种破坏条件下均无明显增加,提示该杂质由原料合成过程带入,需对其进行进一步的研究。
结果分析表明,改进后的方法单个杂质、总杂质及杂质个数均明显增加,提示改进后的方法能更全面的检出杂质,从而更好地控制产品的质量。
猜你喜欢
承德医学院学报(2022年2期)2022-05-23
能源工程(2021年6期)2022-01-06
现代信息科技(2021年21期)2021-05-07
家庭影院技术(2021年2期)2021-03-29
数字海洋与水下攻防(2020年5期)2021-01-04
东坡赤壁诗词(2020年3期)2020-07-04
文萃报·周五版(2020年24期)2020-06-22
启迪与智慧·下旬刊(2019年2期)2019-09-10
中国药房(2018年3期)2018-10-19
教育(2017年38期)2017-09-03
